RNA extraction, probe preparation, and competitive hybridization for transcriptional profiling using Neurospora crassa long-oligomer DNA microarrays
Travis A. Clark1, Julie M. Guilmette1, 2, Daniel Renstrom2, and Jeffrey P. Townsend1
1 Department of Ecology and Evolutionary Biology, Yale University, New Haven, 06520-8106, United States.
2 Department of Molecular and Cell Biology, University of Connecticut, Storrs, 06269-2054, United States.
Fungal Genetics Reports 55:18-28
We developed protocols optimized for the performance of experiments assaying genomic gene expression using Neurospora crassa long-oligomer microarrays. We present methods for sample growth and harvesting, total RNA extraction, poly(A)+ mRNA selection, preparation of NH3-Allyl Cy3/Cy5 labeled probes, and microarray hybridization. The quality of the data obtained with these protocols is demonstrated by the comparative transcriptional profiling of basal and apical zones of vegetative growth of N. crassa.
To further the technology for inference of gene expression levels in Neurospora crassa, whole-genome-spotted oligonucleotide microarrays have been created that contain a total of ~10,500 open reading frames (Dunlap et al. 2007; Tian et al. 2007). These spotted microarrays allow the simultaneous measurement of the abundance of the transcripts from the thousands of genes printed on the arrays through competitive hybridization of labeled cDNA probes (DeRisi and Iyer 1999; Eisen and Brown 1999). The gene expression data generated by the microarrays will help to annotate the N. crassa genome and assign putative functions for unique genes (Dunlap et al. 2007).
Optimal experimental designs for transcriptional profiling using DNA microarrays have been described by Townsend and Taylor (2005). We present in this paper the methods for extracting RNA, purifying poly(A)+ mRNA, cDNA synthesis, Cy dye labeling and microarray hybridization suitable for the long-oligomer Neurospora crassa microarrays (Kasuga et al. 2005; Tian et al. 2007), that are available from the Fungal Genetics Stock Center (FGSC, http://www.fgsc.net).
Methods
Throughout the design and performance of a microarray experiment, keep in mind that the key reagent in transcriptional profiling is the RNA harvested from an organism. This RNA is used as a template for reverse transcription to make cDNA for competitive hybridization against the probes affixed to the microarray. RNA is best extracted from flash-frozen pellets of tissue or culture grown in meticulously maintained “common garden” conditions, in which there are no environmental variations except those mandated by the experiment. It is essential to standardize the protocol to eliminate variation that can alter the gene expression of the sample during the harvesting step (Townsend and Taylor 2005).
Sample Growth and Harvesting: To grow Neurospora, we have inoculated the strain of interest on the center of the petri dish containing a sterilized cellophane membrane overlying solid growth medium. The cellophane membrane permits quick and easy harvesting of the entire sample without contamination of agar. It thus also facilitates accurate measurement of fungal mass for RNA extractions. Use of solid medium may provide closer simulation of growth in natural conditions than cultivation in liquid medium. We used Bird Medium for transcriptional profiling of vegetative growth, rather than Vogel’s Medium, due to the stability of the nutrient components (Metzenberg 2004). The stability presumably leads to more consistent expression of metabolic genes.
When growth had proceeded appropriately for harvesting, we immediately froze the culture by pouring pure liquid nitrogen directly on the plate surface, being careful to ensure that the whole plate was fully frozen. The frozen dish was then immediately placed in the -80 C freezer until harvesting. If performing a profiling study with a spatial component, strips may be cut while the dish is frozen to avoid pulling off the mycelial sheet.
To harvest most easily, the plate was thawed slowly on ice. As soon as mycelium could be easily separated from the cellophane, but while it was still partially frozen, the mycelial sheet was rolled off the cellophane with a sterile microspatula, then placed into a tared sterile 2 ml microcentrifuge tube (with O-ring). The tube was rapidly weighed to get a measurement of the tissue yield, and the sample was then stored at –80 C until ready to extract RNA.
Total RNA purification
We extracted RNA with TRI REAGENT (Molecular Research Center, Inc. Cincinnati, OH). TRI REAGENT is a monophase solution containing phenol and guanidine thiocyanate. It allows for the simultaneous isolation of RNA, DNA and protein from a single sample (Chomczynski 1993; Chomczynski and Sacchi 1987). The frozen mycelial tissue was homogenized in TRI REAGENT, then phase separated by the addition of 1-bromo-3-chloropropane (BCP) or chloroform. The nucleic acid phase was precipitated with isopropanol, washed with 70% ethanol, and solubilized in DEPC treated water. The RNA isolated using this procedure is suitable for applications such as Northern blotting, poly(A)+ mRNA selection, and RT-PCR.
Homogenization: We homogenized frozen tissue in a 7mL Dounce glass tissue grinder that was treated to remove any residue of RNAase, which yielded higher yields of RNA per mg mycelium than use of glass beads or grinding in liquid nitrogen. Fifty to 100 mg of mycelium was homogenized with 1 ml of TRI REAGENT, ensuring that the sample volume never exceeded 10% of the volume of the TRI REAGENT. To further break up tough mycelia, the homogenate was then centrifuged through a Qiagen Qiashredder column for a duration of 2 minutes at 14,000 rcf. The shredded homogenate was incubated for 5 minutes at room temperature to allow for the dissociation of nucleoprotein complexes. In samples with high polysaccharide content (like fungal mycelium), cleaner preparations were found when the sample was centrifuged at 12,000 rcf for 10 minutes at 4 C. This step pelleted debris, polysaccharides and high molecular weight DNA prior to phase separation.
Phase Separation: The supernatant was supplemented with 100 ul of 1-bromo-3-chloropropane and the tube was covered tightly and shaken vigorously for 15 seconds. This solution was incubated for 15 minutes at 25 C and then centrifuged at 12,000 rcf for 15 minutes at 4 C. The cold temperature was essential to separate RNA without residual DNA contamination in the aqueous phase. The aqueous phase was pipetted to a new RNase free microfuge tube, carefully avoiding contamination due to the carryover of the interphase or the organic phase.
Precipitation: The RNA was precipitated by the addition of 250 ul of isopropanol, then 250 ul of high salt precipitation solution (0.8 M Sodium citrate and 1.2 M NaCl). The addition of the high salt precipitation solution to samples with high polysaccharides and proteoglycans resulted in purer RNA (Chomczynski and Mackey 1995). The samples were stored for 10 minutes at 25 C and centrifuged 12,000 rcf for 8 minutes at 4 C to pellet the RNA.
RNA Wash and Solubilization: The supernatant was removed, leaving the RNA pellet intact. The pellet was then washed by vortexing with 1 ml of 75% RNase free ethanol, three times conscutively, to obtain purer RNA. This purity is demonstrated by spectrophometric measurement of thrice-washed samples that yielded an A260/A280 closer to 2.0 (see Table 1) and the A260/A230 between 2.0 and 2.4 (Farrell, 2005). The RNA pellet was then centrifuged at 7,500 rcf for 5 minutes. To remove the residual ethanol, the RNA pellet was allowed to air dry for 3 – 5 min. The RNA pellet was then dissolved in 77 ul of DEPC-treated water by incubating the tube in a 60 C water bath for 15 minutes. For spectrophometric measurement, we found that placement of 2 ul of the RNA sample in 98 ul of phosphate buffer (3 mM Na2HPO4, pH 8.5) gave highly accurate nucleic acid absorbance ratios as compared to other solutions. The UV absorbance measurement of nucleic acids has been shown to significantly vary depending on the pH and ionic strength of the solution used in the measurements (Wilfinger, Mackey, and Chomzynski, 1997). A spectrophotometric ratio of A260/A280 of about 1.8-2.1 indicates a clean preparation without much protein contamination (Farrell, 2005) and was suitable for direct cDNA synthesis or poly(A)+ mRNA selection. Yield ranged from 150 – 500 ug total RNA per 50 – 100 mg of N. crassa mycelium. The RNA samples were stored at -80 C until they were thawed for mRNA purification.
|
|
RNA diluted in DEPC water |
RNA diluted in Phosphate buffer |
||
|
Sample |
A260/A280 |
Conc. (ug/ml) |
A260/A280 |
Conc. (ug/ml) |
|
N. crassa 1 |
1.53 |
553.6 |
1.92 |
364.8 |
|
N. crassa 2 |
1.48 |
497.6 |
1.73 |
338.4 |
|
N. crassa 3 |
1.46 |
519.2 |
1.98 |
297.6 |
|
N. crassa 4 |
1.50 |
444.8 |
1.83 |
296.0 |
Table 1. Comparison of spectrophotometric readings and concentration calculations of RNA diluted in DEPC treated water and phosphate buffer.
Poly(A)+ mRNA purification
Poly(A)+ mRNA may be purified easily using different commercially available kits that contain oligo(dT)-containing resins or columns that retain poly(A)+ mRNA and allow much of the tRNA and rRNA to pass through (reviewed by Nowrousian, 2004). We have had high yields with the Qiagen Oligotex mRNA kit (Qiagen), but find it expensive when multiple arrays are being performed. We found that using the MRC Oligo(dT)-Cellulose columns (Molecular Research Center, Inc. Cincinnati, OH) was a more economical alternative . The columns retained around 90% of poly(A)+ RNA, used solutions and reagents readily prepared from scratch in the lab, and were regenerated with a simple wash of 1% SDS and 75% ethanol. The oligo(dT) columns were reusable at least ten times each. The columns could be fully decontaminated by washing with 0.1 M NaOH and are stable for at least two years when stored at –20 C.
Selection of Poly(A)+ RNA with the MRC Oligo(dT)-Cellulose columns: Affinity chromatography on oligo(dT) columns is the preferred method for large-scale purification (> 25 ug) of poly(A)+ RNA (Sambrook and Russell 2001).We adapted our protocol below from Molecular Research Center (http://www.mrcgene.com/oligo1222.htm) and Sambrook and Russell (2001). RNA samples stored in DEPC treated water were combined to achieve a concentration of 1-4 mg RNA/ml in 600 ul. This RNA solution was incubated at 65 C for 5 minutes and cooled quickly to room temperature. To bring the RNA solution to the proper concentration for the binding of poly(A)+ mRNA to the oligo(dT), 600 ul of 2x binding buffer (1 M LiCl, 100 mM sodium citrate, 0.2% SDS) was added to the pooled RNA sample. Immediately before use, the oligo(dT) columns were prepared by washing with 500 ul of the binding buffer (0.5 M LiCl, 50 mM sodium citrate, 0.1% SDS). The RNA solution was applied to the oligo(dT) column, the elutant was collected, then applied a second time to the column. The column was then washed twice with 1 ml of the binding buffer. The captured poly(A)+ mRNA was then eluted into a RNase free microfuge tube by the addition of 900 ul of elution buffer (1 mM sodium citrate, 0.1% SDS).
To facilitate precipitation of the mRNA, 100 ul of 5 M LiCl and 2-8 ul of Polyacryl Carrier (MRC, included with the columns) was added to the elutant. This solution was mixed briefly, then 1 ml of isopropanol was added to precipitate the mRNA. To pellet precipitated mRNA, the solution was incubated for 5 minutes at room temperature. The precipitate was centrifuged at 12,000 rcf for 5 minutes at 4 C. The pellet of mRNA was then dissolved in DEPC treated water, and an aliquot was taken for spectrophometric measurement. The yield of mRNA per 1 mg of total RNA was typically around 25 – 35 ug. The mRNA solution was then stored at –80 C until it was thawed for reverse transcription. Finally, the columns were washed with 1.5 ml of 1% SDS, followed by 1 ml of 75% ethanol, then stored wetted at –20 C until further use. The manufacturer states that the oligo(dT) columns can be reused at least 10 times. We suspect a greater number of uses are possible, especially when the column is reused on serial, identical samples. If columns become contaminated, they may be stripped with 1.5 ml of 0.1 M NaOH, and then washed with water until the pH of the elutant is less than 7.5.
Preparation of NH3-Allyl Cy3/Cy5 Labeled Probes and Microarray Hybridization
Reverse Transcription and Hydrolysis: cDNA is a product of enzymatic in vitro synthesis requiring high quality template RNA for a successful cDNA preparation. Both total RNA and purified mRNA have been successfully used as templates for the production of labeled cDNA for microarray hybridization. Oligo-dT which binds with the poly(A) tails of mRNA may be used as the sole primer. Alternatively, a mixture of oligo-dT and oligo-dN primer have been used as primers to reverse transcribe mRNA. To help decrease noise in the microarray hybridization caused by errant labeled cDNAs transcribed from abundant tRNA and rRNA sequences (Townsend and Taylor, 2005) we recommend both isolating the mRNA from the total RNA and using oligo-dT alone. To provide a ligand for the Cy dye labeling, amino-allyl dUTP is incorporated into the cDNA along with the dNTPs
For cDNA synthesis, 2 ug of mRNA was mixed with 1 ul of oligo(dT)12-18 primers (Invitrogen) and 2 ul of ArrayControl RNA Spike Mix, and brought to a final volume of 18 ul with DEPC treated water. The mixture was incubated at 70 C for 10 minutes, then it was cooled on ice for 10 minutes. The final volume for the cDNA synthesis reaction was 31.8 ul and contained 10 mM DTT, 500 uM each of dATP, dCTP and dGTP, 200 uM dTTP, 300 uM aminoallyl-dUTP, and 200 U Superscript II (Invitrogen) reverse transcriptase in 1x first strand reaction buffer. After 120 minutes of reverse transcription at 42 C, the reaction was stopped with 10 ul of 1 M NaOH and 10 ul of 0.5 M EDTA. The mix was then incubated at 65 C for 15 minutes. Finally, 25 ul of 1 M HEPES pH 7.5 was added to stabilize the solution.
Buffer Cleanup: To retain the long polymers of cDNA and discard unincorporated nucleotides, the products of reverse transcription were diluted and filtered in a MicroconYM-30 microconcentrator. Typically, an initial 10-fold dilution of the reverse transcription reaction product was followed by a 20-fold concentration. The Microcon YM-30 contains two parts, the removable filter cup and the filtrate collection tube. A filter cup was weighed for each sample and placed back into its collection tube. The cDNA reaction was added to the filter, and then purified water was added to bring the total volume to 500 ul. The samples were initially centrifuged at 12,400 rcf for 8 minutes, the filter cup weighed, and centrifuged again until the weight of the probe was less than 40 mg. At 12,400 rcf, approximately 30 mg passed through the filter per minute. For the second round of dilution and cleanup, the probe was again brought to a total volume of 500 ul and centrifuged at 12,400 rcf until the probe weight was less than 40 mg. A third and last round of dilution and cleanup was performed: once again the total volume of the probe was brought to 500 ul and centrifuged at 12,400 rcf, this time continuing to centrifuge until the probe weight was about 20-25 mg. The filter cup was then inverted into the collection tube and centrifuged at 1,000 rcf for three minutes to collect the concentrated probe.
Cyanine Dye Coupling: NHS dye was bound to cDNA via amino-allyl-dUTP residues by raising the pH to 10–13 with the addition of 8 ul of freshly made and filter sterilized 0.05 M sodium bicarbonate pH 9. The high pH probe was then added to the appropriate NHS Cy dye aliquot (CyDye Post-Labeling Reactive Dye, Amersham Biosciences, Piscataway, NJ) and mixed briefly by pipetting. The coupling reaction was incubated in the dark at 25 C for 75 minutes. The reaction was then quenched by the addition of 15 ul of 4 M hydroxylamine and incubated in the dark at 25 C for an additional 15 minutes. The labeled cDNA was then purified with the QIAquick PCR purification kit (Qiagen). This elution of about 55 ul of purified cyanine-labeled cDNA was stored at 4 C, and used in less than 24 hours.
Hybridization: For each competitive hybridization, the labeled target cDNAs from two samples were used. One cDNA was labeled with Cy3 and the other was labeled with Cy5. We combined the appropriate Cy3 and Cy5 labeled paired samples in a Microcon YM-30 microconcentrator. The dually labeled cDNA was centrifuged at 12,400 rcf until its weight was 25 mg. The filter cup was then inverted and spun at 1,000 rcf for three minutes, and the elutant was brought to a total volume of 20 ul with purified water. We added 0.375 ul of poly[A] (Polyadenylic acid potassium salt, Sigma-Aldrich, MO) to block nonspecific binding to probes by the poly-T tails of the cDNA. Next, 3 ul of 20x SSC and 0.5 ul of 1 M HEPES pH 7.0 were added. This blocked and buffered target cDNA mix was then filtered of dust or residues with a wetted (10 ul ddH2O) Millipore Ultrafree-MC 0.45 um filter. To the filtered mix we added 0.45 ul of 10% SDS. We then boiled it at 100 C for 2 minutes to denature the nucleic acids. This denatured target cDNA was then cooled to 25 C for 15 minutes. We have found that hybridizations using labeled target at temperatures above room temperature can result in high background fluorescence. A blocked microarray slide (see included blocking protocol) was set in a hybridization chamber (Corning 2551, Corning, NY). To keep the slide stable within the chamber, we deposited drops of 3x SSC on the underside of the slide, allowing them to adsorb to the slide corners and the chamber bottom. To prevent dehydration of the labeled cDNA solution from beneath the coverslips, 40 ul of 3x SSC was added to each of the hybridization chamber wells. A coverslip (LifterSlips were very convenient for this purpose) was cleaned with ethanol and then very carefully placed over the printed microarray. The labeled cDNA mix was then injected at the corners of the coverslip. This step was done quickly, but precisely, to avoid non-uniform cooling of the probe mix that can cause uneven background, and to avoid forming bubbles under the coverslip. The hybridization chamber was sealed, carefully kept level, and then placed level in a 53 C waterbath. Incubation occurred at 53 C for 12 hours to reach equilibrium.
Array Wash: Hybridized microarray slides were washed by repeated plunging (at least twenty times) in a solution of 387 ml of purified water, 12 ml of 20x SSC, and 1 ml of 10% SDS, and rinsed by repeated plunging in a solution of 399 ml of purified water and 1 ml of 20x SSC. Residual water was rapidly removed from the slide by centrifugation at 450 rcf for 1 minute. The array was scanned as soon as possible; if there was any delay, the array was stored in the dark for no more than 2 hours.
Data Acquisition and Analysis: Fluorescent DNA bound to the microarray was detected with a GenePix 4000 microarray scanner (Axon Instruments, Foster City, CA). The GenePix Pro 6.0 software package was used to locate spots in the microarray. The GenePix Array List (GAL) file used to grid and identify spots on the N. crassa array may be downloaded from the Filamentous Fungal Database (http://www.yale.edu/townsend/Links/ffdatabase/downloads.html).
Blocking the microarrays: The N. crassa long-oligomer microarrays available from the FGSC (http://www.fgsc.net), were printed on Corning UltraGAPS gamma amino propyl silane coated slides and were processed prior to use. The microarray slides were first rehydrated by placing in a room temperature humid chamber for around 5 minutes, until the printed spots glistened, but did not congeal. The slide was then snap-dried by placing it on an 80 C heating block for one minute. The oligonucleotides on the slide were then crosslinked by treating the slide with 600 mJ of UV. A 500 ml graduated cylinder was cleaned and acetone-washed, then 335 ml 1-methyl-2-polypyrolidinone and 6 g succinic anhydride were added and the mix was stirred. Immediately upon dissolution, 15 ml of 1 M sodium borate (pH 8.0) was added, allowed to be dispersed evenly into the solution, and immediately poured into a 500 ml Wheaton glass tank. The crosslinked slides were first washed by plunging five times in the solution and then the Wheaton tank containing the slide rack and the wash solution was shaken gently for 15 minutes. After shaking, the slide rack was immediately plunged 5 times in boiling water, and then the slides were let to sit for 2 minutes in the boiling water. They were then plunged 5 times in 95% ethanol (not 100% or diluted 100% ethanol). Finally, the slides were spun dry for 15 seconds at 1000 rcf and stored in a slide container in a desiccator until use (< 1 year).
Experimental Verification of Protocols
To verify the protocols presented, samples were taken from spatially discrete regions of the mycelium of N. crassa using a defined and repeatable growth protocol (CG Tian, http://www.yale.edu/townsend/Links/ffdatabase/assets/Large Plate Protocol.pdf). An N. crassa long-oligomer microarray was used to compare the apical, leading edge of the mycelium and a region of penultimate mycelial growth.
One strain of N. crassa
was used throughout the experiment: FGSC#2489 (Fungal Genetics Stock Center,
Kansas City, MO). Solid 1.5% agar Bird medium with 1.8% glucose as a carbon
source was used for all stages of the experiment (Metzenberg 2004). A 245 mm
square BioAssay Dish (Corning, Corning, NY) was poured and covered with a sheet
of wetted and sterilized cellophane (Fisher Scientific). 100 ul of conidial
solution then was spread over the cellophane surface. To avoid the temperature
variation typical to room temperature growth, ambient temperature was
maintained at a climate-controlled 23 C, under constant light, for 36
hours. A 1 x 21 cm strip was cut 2 cm from the edge of the first plate, where
conidia growth was minimal. This strip was used for inoculation of a second 245
mm square dish (also overlain with cellophane), by placing the excised
cellophane strip vertically across the center of the plate. Strip-inoculated
plates were grown under constant light at 23 C for 30 hours. The
mycelia were allowed to grow to 3 cm from the edge of the plate. The
development of the mycelia was halted through flash freezing with clean liquid
nitrogen. There was minimal disruption of the spatial structure of the mycelia
from the vortex created by the application of the liquid nitrogen. Mycelia were
then harvested by first cutting the cellophane into 1 x 7 cm strips followed by
cutting horizontally into 3 sections, all approximately 7-7.5 cm long. Each
strip was placed into one microfuge tube and stored at –80 C until
extraction. The mycelium strips were homogenized in the presence of ~3 mg of 0.5
mm Zirconia-silica beads (Biospec Products Inc., Bartlesville, OK) and 1 ml TRI
REAGENT (Molecular Research Center Inc., Cinncinnati, OH) for 5 min using the
Disruptor Genie (Scientific Industries Inc., Bohemia, NY). Other than these
exceptions, the samples were processed by the well-tested protocols described in
detail at the beginning of this paper.
The RNA
harvested for microarray analysis in this study was taken from neighboring
mycelial strips that are not presumed to have fairly limited differences in gene
expression. The 1 x 7 cm mycelial strip containing the growing edge of the
mycelium hereafter referred to as the “apical” strip was harvested and the mRNA
was labeled with Cy5. and the first 1 x 7 cm mycelial strip cut after the
inoculation point at the center of the plate, hereafter referred to as the
“penultimate” strip was harvested and the mRNA was labelled with Cy3.
Data Acquisition and Analysis
A two-channel color image of array fluorescence was acquired with an Axon GenePix 4000B microarray scanner and the GenePix 4000 software package (Axon Instruments, Foster City, CA). The automated GenePix spot-finding algorithm was used, complemented by manual image inspection in order to best capture of reporter spot size, shape and quality. A global mean-by-mean normalization of well-measured Cy3 and Cy5 signal intensities was carried out using an Excel template spreadsheet (http://www.yale.edu/townsend/Links/ffdatabase/downloads.html). Spots were excluded from further analysis if Cy3 and/or Cy5 signal intensities were within two standard deviations (SD) of the distribution of background pixel intensities (Townsend and Taylor 2005).
Complete experimental designs should be analyzed by robust ANOVA or Bayesian statistical methodologies (Townsend and Hartl 2002; Wolfinger et al. 2001) incorporating dye-swapping and significant replication (Townsend and Taylor 2005). For this technical verification, only those ORFs spotted on the array showing a strict two-fold increase or decrease in ratio intensities are reported based on background-corrected normalized signal intensity ratios. ORFs conforming to the strict two-fold analysis were subsequently categorized by comparison to a spreadsheet obtained from the developers of the Functional Catalogue (Ruepp et al. 2004). The Functional Catalogue is accessible from the Munich Information Center for Protein Sciences (MIPS). Other annotation information was obtained from The InterPro Database (Apweiler et al. 2001; Mulder et al. 2003), Enzyme Commission numbers (http://www.chem.qmul.ac.uk/ iubmb/enzyme), and the KEGG database (Kanehisa et al. 2006).
Results and Discussion
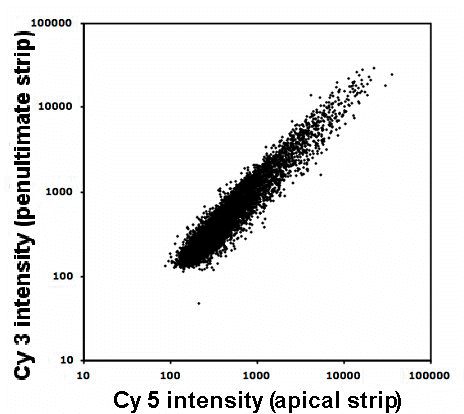
Following manual inspection and normalization, mean spot intensities for 8193 ORFs (open reading frames) were at least two SD above background levels and contained less than 2% saturated pixels. Spots passing these criteria were termed “strict pass points.” The slope of the plotted Cy3 and Cy5 log values for these strict pass points would optimally be 1.00. Figure 1 shows the linear regression of the strict pass points with a slope of 1.093 and a correlation coefficient of 0.879, consistent with a high quality array (Townsend and Taylor 2005). The average signal intensities for Cy3 and Cy5 for those strict pass points were 686 and 654 respectively. The normalization template provided output in the form of adjusted ratios between signal intensities for the Cy5 (635nm) and Cy3 (532nm) channels. A total of 52 ORFs were termed “abundant” in the apical strip compared to the penultimate strip (i.e. the ratio was [greater than 2.0/less than 0.5]), while 76 ORFs were termed “abundant” in the penultimate strip compared to the apical strip (i.e. the ratio was [greater than 2.0/less than 0.5]).
Figure 1. The linear regression of the Cy3 and Cy5 log intensity values for spots passing a “strict pass” criterion. The strict pass criterion for a well-measured spot required that the background-subtracted average foreground intensity had to exceed three standard deviations of the background pixel intensities.
Use of MIPS FunCat (Ruepp et al. 2004) provided functional information for many of the differentially expressed ORFs (See Table 2 and Table 3). Comparison between the transcription/translation categories has yielded the following results. ORFs abundantly expressed in the apical strip were commonly involved in rRNA processing, while those slightly expressed in the apical strip were commonly involved in transcriptional activity or regulation. Out of the 52 ORFs abundantly expressed in the apical strip, 17 (32.7%) were found to have no functional categorical information available. The remaining ORFs from the apical strip were placed in functional categories based on annotation using the four aforementioned protein database tools. In total, the 35 abundantly expressed ORFs were placed in eight categories: transcription/translation/DNA repair, cell differentiation/control, carbon metabolism, amino acid/nucleotide metabolism, cell wall growth, respiration/oxidative phosphorylation, transport/regulation, and protein modification. The categories that contained the most ORFs were transport/regulation and cell wall growth, with eight and seven ORFs respectively. The categories with the fewest abundantly expressed ORFs were carbon metabolism and cell differentiation/control, each containing two. Most of the ORFs categorically placed were classified due solely to sequence or domain homology with known proteins from other organisms.
In the penultimate strip, 32 of the 76 ORFs (42.1%) abundantly expressed were found to have no functional categorical information available. The remaining 44 ORFs fell within one of seven major categories: carbon/nitrogen utilization, cellular transport, cell wall/cell membrane structure, transcription/translation, cytoskeletal structure, oxidative phosphorylation and protein modification. Most of these ORFs were also classified based on sequence/domain homology between previously encountered genes in other organisms. NCU03753.1 has previously been associated with the gene grg-1 in N. crassa, commonly known as clock-controlled gene 1 (ccg-1; Arpaia et al., 1995; Kothe and Free, 1998). The signal intensity for grg-1 was 3.2 times more abundant in the penultimate strip compared to the apical strip. Due to the decrease in medium glucose concentration inward from the vegetative growth front, we expected to see an expression difference in NCU03753.1/grg-1, the glucose-repressible gene-1 (synonym clock-controlled gene 1, ccg-1). The grg-1 gene was three-fold more abundant in the penultimate strip indicating it functions in older mycelia, most likely due to the decreased amount of glucose left in the growth medium at this position. As the name indicates this gene is repressed by addition of glucose to the medium (Davis 2000; Wang et al. 1994). A study by Tey et al. (2005) also found that grg-1 had increased expression in areas farther from the hyphal apical tip.
Two ORFs NCU08077.1 and NCU02887.1, abundant in the apical strip have homology to proteins involved in calcium ion binding. During hyphal extension Ca+2 ions are concentrated at the apical tip. Following elongation it is assumed that a set of Ca+2 binding proteins are recruited to bind free Ca+2 and redistribute it to avoid increasing free Ca+2 concentrations which can prove to be toxic (Davis, 2000). The abundance of two different Ca+2 binding proteins in the older hyphal growth is consistent with the hypothesized calcium use in hyphal elongation.
Unlike many other fungi, Neurospora crassa is not known to exhibit pathogenicity in plants (Borkovich et al. 2004; Galagan et al. 2003). Interestingly, two ORFs more abundant in the growing apical strip are homologous to genes that are associated with pathogenicity in other fungal species. NCU08038.1 has been classified as a protein in the Egh16/Egh16H gene family that have been shown to be upregulated during infection of plants (Grell, Mouritzen, and Giese 2003). NCU09663.1 has been more specifically classified as a cutinase precursor (Borkovich et al. 2004). These two ORFs may have at one time encoded these proteins for pathogenic purposes in an ancestor of N. crassa, but is more likely that these proteins have general growth associated functions unrelated to pathogenicity in N. crassa. Similarly the protein associated with NCU08038.1 may play a cell differentiation role in apical cells in an pathogenic fungi or even a pathogenic fungus ancestral to N. crassa.
The comparison of the results found in this study with those from published studies suggest that the protocols presented here are appropriate for experiments transcriptionally profiling N. crassa using the long-oligomer microarrays. We hope by presenting these protocols other laboratories will have similar success.
|
Functional Category |
|
|
Locus # |
|
Fold Change |
|
|
|
|
|
|
|
|
|
|
Transcription/Translation/DNA repair |
|
|
|
|
||
|
|
Eukaryotic/archaeal ribosomal protein S3 |
NCU00489.1 |
|
2.08 |
||
|
|
rRNA synthesis/rRNA processing; Fibrillarin |
NCU03702.1 |
|
2.08 |
||
|
|
related to methyltransferase involved in pre-rRNA cleavage |
NCU05293.1 |
|
2.02 |
||
|
|
rRNA processing; ATP-dependent helicase |
NCU05782.1 |
|
2.00 |
||
|
|
ATP-dependent helicase, rRNA/tRNA/mRNA processing |
NCU07839.1 |
|
2.00 |
||
|
|
|
|
|
|
|
|
|
Cell Differentiation/Control |
|
|
|
|
|
|
|
|
cell type diff; hyphae formation; probable gEgh 16 protein |
NCU08038.1 |
|
2.65 |
||
|
|
dyp-type peroxidase |
|
NCU09210.1 |
|
2.06 |
|
|
|
related to YSC84 - protein involved in the organization of the actin cytoskeleton |
NCU05881.2 |
|
2.19 |
||
|
|
|
|
|
|
|
|
|
Carbon Metabolism |
|
|
|
|
|
|
|
|
dehydrogenase; possibly related to alcohols/glucose |
NCU04592.1 |
|
2.58 |
||
|
|
aldehyde dehydrogenase [NAD(P)] |
|
NCU09648.1 |
|
2.17 |
|
|
|
related to RAY38 protein |
|
NCU00225.2 |
|
2.65 |
|
|
|
|
|
|
|
|
|
|
Amino Acid/Nucleotide Metabolism |
|
|
|
|
||
|
|
glycine/serine/threonine metab. |
|
NCU09798.1 |
|
2.69 |
|
|
|
related to 5`-nucleotidase precursor |
NCU09659.1 |
|
2.08 |
||
|
|
Methylthioadenosine phosphorylase |
|
NCU03963.1 |
|
2.00 |
|
|
|
|
|
|
|
|
|
|
Cell Wall / Cell Membrane Growth |
|
|
|
|
||
|
|
phospholipid biosyth/degradation; glycerolipid metabolism |
NCU04475.1 |
|
3.21 |
||
|
|
related to cellulose synthase |
|
NCU08226.1 |
|
2.74 |
|
|
|
lipid, fatty acid and isoprenoid biosynthesis |
NCU07937.1 |
|
2.24 |
||
|
|
cell wall biogen. (starch synthase/a-1,3-glucan synthase) |
NCU08132.1 |
|
2.18 |
||
|
|
Glycoside hydrolase, family 61 |
|
NCU02344.1 |
|
2.17 |
|
|
|
triacylglycerol lipase |
|
NCU03639.1 |
|
2.15 |
|
|
|
related to acetyl xylan esterase II precursor |
|
NCU09663.1 |
|
2.01 |
|
|
|
|
|
|
|
|
|
|
Respiration/Oxidative Phosphorylation |
|
|
|
|
||
|
|
Cytochrome P450 |
|
|
NCU05376.1 |
|
2.37 |
|
|
aerobic respiration; NADH2 dehydrogenase (ubiquinone) |
NCU10873.2 |
|
2.06 |
||
|
|
NADH2 dehydrogenase (ubiquinone) |
|
NCU02280.1 |
|
2.06 |
|
|
|
|
|
|
|
|
|
|
Transport/Regulation |
|
|
|
|
|
|
|
|
Patatin |
|
|
NCU00381.1 |
|
2.39 |
|
|
Regulator of G protein |
|
NCU09415.1 |
|
2.09 |
|
|
|
ER to Golgi transport, SEC7-like domain |
NCU07658.1 |
|
2.07 |
||
|
|
vesicular transport, exocytosis, Synaptojanin |
NCU00896.1 |
|
2.05 |
||
|
|
Exocytosis |
|
|
NCU09432.1 |
|
2.03 |
|
|
proton driven symporter, nucleoside transporter |
NCU08148.1 |
|
2.02 |
||
|
|
peptide transport (rel. to PTR2), cellular import |
NCU08738.1 |
|
2.01 |
||
|
|
non-vesicular cellular import, amino acid permease |
NCU05168.1 |
|
2.01 |
||
|
|
|
|
|
|
|
|
|
Protein Modification |
|
|
|
|
|
|
|
|
Peptidase M, neutral zinc metallopeptidases |
NCU06548.1 |
|
2.52 |
||
|
|
protein degradation; Tripeptidyl-peptidase I |
NCU08418.1 |
|
2.21 |
||
|
|
Peptidase M12B, ADAM/reprolysin, protease ADM-B |
NCU06834.1 |
|
2.10 |
||
|
|
related to hxB protein |
|
NCU03011.1 |
|
2.00 |
|
|
|
|
|
|
|
|
|
|
Unknown |
|
|
|
|
||
|
|
conserved hypothetical protein |
|
NCU00265.1 |
|
3.42 |
|
|
|
conserved hypothetical protein |
|
NCU06227.2 |
|
2.17 |
|
|
|
conserved hypothetical protein, secretion |
|
NCU00092.1 |
|
2.10 |
|
|
|
conserved hypothetical protein, mitochondrion |
|
NCU05202.2 |
|
2.10 |
|
Table 2. Genes abundantly expressed in the apical sample of the mycelium.
|
Functional Category |
|
|
Locus # |
|
Fold Change |
|
|
|
|
|
|
|
|
|
|
Transcription/Translation |
|
|
|
|
|
|
|
|
ribosomal proteins, Ribosomal protein S21e |
NCU08627.1 |
|
2.00 |
||
|
|
Fungal transcriptional regulatory protein |
NCU08049.1 |
|
2.03 |
||
|
|
related to origin recognition complex subunit 4 |
NCU08317.1 |
|
2.04 |
||
|
|
aminoacyl-tRNA-synthetases |
|
NCU00466.1 |
|
2.05 |
|
|
|
related to Hsp90 co-chaperone Cdc37 |
|
NCU00472.1 |
|
2.11 |
|
|
|
probable DNA replication licensing factor (nimQ) |
NCU04327.1 |
|
2.13 |
||
|
|
Histone-fold/TFIID-TAF/NF-Y |
|
NCU02161.1 |
|
2.23 |
|
|
|
Tudor domain/Staph. nuclease subtype; DNA binding |
NCU02134.1 |
|
2.45 |
||
|
|
ATP-dependent DNA ligase |
|
NCU01365.1 |
|
2.51 |
|
|
|
|
|
|
|
|
|
|
Cellular Transport |
|
|
|
|
|
|
|
|
Lipocalin-related protein |
|
NCU00730.1 |
|
2.04 |
|
|
|
G-protein mediated signal transduction |
|
NCU07173.1 |
|
2.06 |
|
|
|
Transport protein particle (TRAPP) component, Bet3 |
NCU05796.1 |
|
2.07 |
||
|
|
TONB Box |
|
|
NCU09765.1 |
|
2.08 |
|
|
related to endosomal Vps protein complex subunit |
NCU01791.1 |
|
2.11 |
||
|
|
Calcium-binding EF-hand |
|
NCU08077.1 |
|
2.14 |
|
|
|
Autophagocytosis associated protein |
|
NCU01955.1 |
|
2.28 |
|
|
|
homeostasis of metal ions (Na, K, Ca etc.) |
NCU02887.1 |
|
2.43 |
||
|
|
cellular import |
|
|
NCU07820.1 |
|
2.60 |
|
|
major facilitator superfamily, small solute transport |
NCU08428.1 |
|
2.65 |
||
|
|
probable glucose transport protein |
|
NCU08180.1 |
|
2.91 |
|
|
|
|
|
|
|
|
|
|
Cell Wall / Cell Membrane Growth |
|
|
|
|
|
|
|
|
Fatty acid synthase |
|
NCU07307.1 |
|
2.01 |
|
|
|
Isopentenyl-diphosphate delta-isomerase |
NCU05014.1 |
|
2.24 |
||
|
|
rel. to ECM15 protein, cell wall biogenesis and architecture |
NCU06201.1 |
|
2.26 |
||
|
|
biosynthesis of phenylpropanoids (including lignin) |
NCU00677.1 |
|
2.44 |
||
|
|
3-oxoacyl-[acyl-carrier-protein] reductase |
NCU04791.1 |
|
2.60 |
||
|
|
related to cellodextrin-phosphorylase |
|
NCU09425.1 |
|
2.63 |
|
|
|
|
|
|
|
|
|
|
Cytoskeletal Structure |
|
|
|
|
|
|
|
|
mitotic cell cycle and cell cycle control, actin/actin-like |
NCU02555.1 |
|
2.02 |
||
|
|
Spectrin repeat |
|
|
NCU01491.1 |
|
2.23 |
|
|
Spectrin repeat domain |
|
NCU01273.1 |
|
2.71 |
|
|
|
|
|
|
|
|
|
|
Carbon/Nitrogen Utilization |
|
|
|
|
|
|
|
|
nitrogen and sulfur utilization, Cyanate lyase |
NCU01258.1 |
|
2.02 |
||
|
|
degradation of glutamate, Glu/Leu/Phe/Val dehydrogenase |
NCU00461.1 |
|
2.03 |
||
|
|
polysaccharide degradation, b-glucosidase |
NCU00130.1 |
|
2.09 |
||
|
|
Glycoside hydrolase, family 76 |
|
NCU09937.1 |
|
2.10 |
|
|
|
Aldose 1-epimerase like |
|
NCU02786.1 |
|
2.49 |
|
|
|
glucose-repressible gene-1 protein (grg-1, aka ccg-1) |
NCU03753.1 |
|
3.21 |
||
|
|
|
|
|
|
|
|
|
Protein Modification |
|
|
|
|
|
|
|
|
Protein kinase |
|
|
NCU01113.1 |
|
2.05 |
|
|
Dual specificity protein phosphatase |
|
NCU05049.1 |
|
2.17 |
|
|
|
Serine/threonine protein kinase |
|
NCU00682.1 |
|
2.33 |
|
|
|
dihydrodipicolinate synthase, lysine biosynthesis |
NCU05037.1 |
|
2.39 |
||
|
|
ubiquitin-dependent proteolysis |
|
NCU04328.1 |
|
2.67 |
|
|
|
|
|
|
|
|
|
|
Oxidative Phosphorylation |
|
|
|
|
|
|
|
|
Cytochrome c heme-binding site |
|
NCU08241.1 |
|
2.17 |
|
|
|
pantetheine-phosphate adenylyltransferase |
NCU06029.1 |
|
2.27 |
||
|
|
|
|
|
|
|
|
|
Multiple Categories |
|
|
|
|
|
|
|
|
Ankyrin |
|
|
NCU09991.1 |
|
2.08 |
|
|
|
|
|
|
|
|
|
Unknown |
|
|
|
|
|
|
|
|
hypothetical protein |
|
NCU04477.1 |
|
2.87 |
|
|
|
hypothetical protein |
|
NCU05327.1 |
|
2.65 |
|
|
|
conserved hypothetical protein, secretory |
|
NCU06570.1 |
|
2.40 |
|
|
|
conserved hypothetical protein, mitochondrion |
|
NCU04649.1 |
|
2.35 |
|
|
|
conserved hypothetical protein |
|
NCU09100.1 |
|
2.34 |
|
|
|
conserved hypothetical protein, secretory |
|
NCU06682.1 |
|
2.24 |
|
|
|
conserved hypothetical protein, secretory |
|
NCU05768.1 |
|
2.24 |
|
|
|
conserved hypothetical protein, secretory |
|
NCU07449.1 |
|
2.23 |
|
|
|
conserved hypothetical protein, mitochondrion |
|
NCU01011.1 |
|
2.20 |
|
|
|
conserved hypothetical protein |
|
NCU00864.1 |
|
2.14 |
|
|
|
conserved hypothetical protein, secretory |
|
NCU09029.1 |
|
2.14 |
|
Table 3. Genes abundantly expressed in the penultimate sample of the mycelium.
Acknowledgements
This work was supported by funding awarded to JPT from a National Institutes of Health multi-institutional program project grant (GM068087).
References
Arpaia, G., J. J. Loros, J. C. Dunlap, G. Morelli, and G. Macino. 1995. Light induction of the clock-controlled gene ccg-1 is not transduced through the circadian clock in Neurospora crassa. Mol Gen Genet 247:157-63.
Apweiler, R., T. K. Attwood, A. Bairoch, A. Bateman, E. Birney et al., 2001. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Research 29: 37-40.
Borkovich, K. A., L. A. Alex, O. Yarden, M. Freitag, G. E. Turner et al., 2004. Lessons from the genome sequence of Neurospora crassa: Tracing the path from genomic blueprint to multicellular organism. Microbiology and Molecular Biology Reviews 68: 1-108.
Chomczynski, P., 1993. A Reagent for the Single-Step Simultaneous Isolation of RNA, DNA and Proteins from Cell and Tissue Samples. Biotechniques 15: 532-&.
Chomczynski, P., and K. Mackey, 1995. Modification of the Tri-Reagent(Tm) Procedure for Isolation of Rna from Polysaccharide-Rich and Proteoglycan-Rich Sources. Biotechniques 19: 942-945.
Chomczynski, P., and N. Sacchi, 1987. Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate Phenol Chloroform Extraction. Analytical Biochemistry 162: 156-159.
Davis, R. H., 2000. Neurospora: Contributions of a Model Organism. Oxford University Press, Oxford.
Derisi, J. L., and V. R. Iyer, 1999. Genomics and array technology Current Opinion in Oncology 11: 76-79.
Dunlap, J. C., Borkovich K. A., Henn M. R., Turner G. E., Sachs M. S. et al., 2007. Enabling a community to dissect an organism: overview of the Neurospora functional genomics project. Advances in Genetics 57: 49-96.
Eisen, M. B., and P. O. Brown, 1999. DNA arrays for analysis of gene expression. Methods in Enzymology 303: 179-205.
Galagan, J. E., S. E. Calvo, K. A. Borkovich, E. U. Selker, N. D. Read et al., 2003. The genome sequence of the filamentous fungus Neurospora crassa. Nature 422: 859-868.
Grell, M. N., P. Mouritzen, and H. Giese, 2003. A Blumeria graminis gene family encoding proteins with a C-terminal variable region with homologues in pathogenic fungi. Gene 311: 181-192.
Kanehisa, M., S. Goto, M. Hattori, K. F. Aoki-Kinoshita, M. Itoh et al., 2006. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Research 34: D354-D357.
Kasuga, T., J. P. Townsend, C. G. Tian, L. B. Gilbert, G. Mannhaupt et al., 2005. Long-oligomer microarray profiling in Neurospora crassa reveals the transcriptional program underlying biochemical and physiological events of conidial germination. Nucleic Acids Research 33: 6469-6485.
Kothe, G. O., and S. J. Free. 1998. Calcineurin subunit B is required for normal vegetative growth in Neurospora crassa. Fungal Genet Biol 23:248-58.
Metzenberg, R. L., 2004. Bird Medium: an alternative to Vogel Medium. . Fungal Genetics Newsletter 51: 19-20.
Mulder, N. J., R. Apweiler, T. K. Attwood, A. Bairoch, D. Barrell et al., 2003. The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Research 31: 315-318.
Ruepp, A., A. Zollner, D. Maier, K. Albermann, J. Hani et al., 2004. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Research 32: 5539-5545.
Sambrook, J., and D. W. Russell, 2001. Selection of poly(A)+ mRNA by oligo(dT)-cellulose chromatography. in Molecular Cloning: A Laboratory Manual. CSH Laboratory Press, Cold Spring Harbor, NY.
Tenney, K., I. Hunt, J. Sweigard, J. I. Pounder, C. Mcclain et al., 2000. hex-1, a gene unique to filamentous fungi, encodes the major protein of the Woronin body and functions as a plug for septal pores. Fungal Genetics and Biology 31: 205-217.
Tey, W. K., A. J. North, J. L. Reyes, Y. F. Lu and G. Jedd, 2005. Polarized gene expression determines Woronin body formation at the leading edge of the fungal colony. Molecular Biology of the Cell 16: 2651-2659.
Tian C., T. Kasuga, M. S. Sachs, and N. L. Glass, 2007. Transcriptional profiling of cross pathway control in Neurospora crassa and comparative analysis of the Gcn4 and CPC1 regulons. Eukaryotic Cell 6: 1018-1029.
Townsend, J. P., and D. L. Hartl, 2002. Bayesian analysis of gene expression levels: statistical quantification of relative mRNA level across multiple strains or treatments. Genome Biology. Genome Biology research0071.0071–0071.0016.
Townsend, J. P., and J. W. Taylor, 2005. Designing experiments using spotted microarrays to detect gene regulation differences within and among species. Methods in Enzymology 395: 597-617.
Wang, Z., M. Deak and S. J. Free, 1994. A cis-acting region required for the regulated expression of grg-1, a Neurospora glucose-repressible gene. Two regulatory sites (CRE and NRS) are required to repress grg-1 expression. Journal of Molecular Biology 237: 65-74.
Wolfinger, R. D., G. Gibson, E. D. Wolfinger, L. Bennett, H. Hamadeh et al., 2001. Assessing gene significance from cDNA microarray expression data via mixed models. Journal of Computational Biology 8: 325-637.