A procedure for parallel purification of
four stress-related Neurospora proteins in their native state.
M. Kapoor. Department of Biological Sciences, University of Calgary, Calgary, Alberta, Canada
Fungal Genetics Reports 65:8-11
A procedure is described for isolation and purification of four high-molecular mass stress-responsive proteins from the same starting material.
Proteins are the major components determining cellular growth, metabolism, regulation, repair, homeostatic mechanisms, differentiation, organogenesis, response to environmental factors and ultimately survival. Extensive protein networks form the basis of interlocking metabolic pathways and their regulatory circuits in the eukaryotic and prokaryotic cells, involving transient interactions among a large, overwhelming array of actors. Availability of purified proteins in their native state is critical on account of the universal
interest in unraveling how proteins function in their endogenous intracellular environment.
With the rapid progress in genomics and availability of an ever increasing number of crystal structures, an understanding of the molecular basis of interactions of proteins with ligands, substrates and regulatory molecules is achievable. Structural studies, for instance X-ray crystallographic analysis, depend on homogeneous preparations of proteins. The use of heterologous expression systems with bacteria as host cells is a well established methodology. Other host systems include yeast, insect cells and plant cells. The technology for cloning and expression of proteins, vectors with selectable markers, enhancers, trafficking signals and a range of facile protocols for over-expression are also readily available. However, often it is feasible to express only individual domains of large multi-domain proteins efficiently in heterologous systems. Even when the entire polypeptide can be expressed it is not always possible for the host cell to perform the necessary post-translational modifications.
Furthermore, it is often difficult to obtain sufficient material— tissues/cells—to enable recovery of requisite quantities of the critical protein for physico-chemical analyses. If the source organism is easy to cultivate and adequate quantities of starting material can be acquired in a relatively short time, isolation and purification of the target protein(s) in the native state presents the best opportunity for insightful structural studies. On account of its rapid growth and simple nutrient requirements Neurospora is the ideal organism for acquisition of purified proteins.
The following is a description of a procedure developed in my laboratory for isolation of high- molecular-mass stress-related Neurospora proteins (Fig. 1): the molecular chaperones nHsp70, nHsp80, heat-induced peroxidase (HI-per) and BrnE (Band running next to Hsp Eighty) subsequently identified as the oxidative stress-responsive, cobalamin-independent methionine synthase (MetS). This multiplex protocol results in the recovery of four proteins from the same starting material: common initial steps are followed by fractionation schemes specific to individual proteins, thereby generating near-homogeneous preparations. The general methodology entails standard protein precipitation, ion-exchange, hydrophobic- and affinity- chromatography steps (1). The overall yield of the final products is in milligram quantities, sufficient for identification by MALDI-TOF MS analysis, examination of physico-chemical properties and structural studies. Details of the protocol are outlined in the accompanying flow chart.
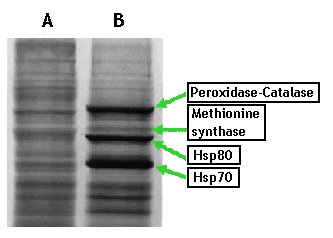
Figure 1. Autoradiographs depicting SDS-PAGE profile of non-shocked (A) and heat-shocked mycelial cells (B), grown for 14 h and labeled with [35S] methionine for 1 h during heat-shock treatment.
Experimental
1. Five grams of lyophilized, heat-shocked Neurospora crassa mycelium is suspended in 40 mL of Buffer A (50 mM Tris-HCl, pH 7.5- 10 mM β-mercaptoethanol- 0.01 mM EDTA) containing 1 mM PMSF (phenylmethanesulfonyl fluoride) or ‘Complete’ protease inhibitor cocktail (Roche) and stirred in the cold room for 30 min. The slurry is homogenized using a Potter-Elvehjem glass homogenizer and centrifuged for 20 min, at 12000g in a refrigerated centrifuge at 4oC. The pelleted material is discarded.
1.1 The supernatant is subjected to ammonium sulfate (AS) fractionation; the pellet from the 40 to 80% saturation fraction is collected by centrifugation and re-suspended gently in ~5 mL of 20 mM Tris-HCl (pH 7.5) - 20 mM NaCl - 10 mM MgCl2 (Buffer B) and dialyzed against five 2-L changes of the same buffer to remove residual AS.
1.2 The dialyzed sample is next loaded on a 15 mm x 30 cm DEAE cellulose (or Sephadex) column equilibrated against buffer A. The column is washed with 60 mL of the loading buffer and the wash solution is subjected to one-dimensional SDS-polyacrylamide gel (10%) electrophoresis (2) to detect the target proteins. The flow-through fractions contain BrnE (methionine synthase) and the high-molecular mass heat-induced peroxidase, while nHsp70 and nHsp80 are retained in the column matrix. Hereafter, the procedure bifurcates.
2. Separation and
purification of nHsp70 and nHsp80
2.1 Next, the bound and unbound fractions from 1.2 are processed by separate chromatography steps. The bound fraction—containing nHsp70 and nHsp80—is eluted with a 0 to 1 M NaCl gradient in Buffer B. Aliquots from fractions of the eluant (~2.5-mL) are inspected by electrophoresis in SDS-polyacrylamide gels for the presence of nHsp70 and nHsp80. The pooled fraction containing these two proteins is applied directly to a 14-mL Cibacron Blue-agarose 3GA (Sigma) column, pre-equilibrated against 20 mM Tris-HCl, pH 7.5 - 20 mM NaCl - 10 mM MgCl2. Both proteins bind to this column; elution is performed employing a 0 to 1.75 M NaCl linear gradient, the emerging fractions being scrutinized by SDS-PAGE. The two molecular chaperones are seen to copurify up to this point.
2.1.1 Purification of nHsp70: Enriched fractions from the Cibacron-blue column are pooled and applied directly to a 1-mL ATP agarose (Sigma) column, pre-equilibrated against the same buffer. The column is washed with buffer B containing 0.6 M NaCl. At this stage nHsp70 and nHsp80 are separated from each other: the latter is recovered in the low-salt flow-through fraction while the former remains attached to the matrix. The bound nHsp70 is eluted with a small volume of buffer B containing 5 mM ATP and the eluted solution is concentrated using 30-kD cutoff filter units. This series of steps yields virtually pure nHsp70 (Fig. 2) its identity being verified by MALDI-TOF mass spectrometry and Western blotting using Neurospora Hsp70-specific polyclonal IgG.
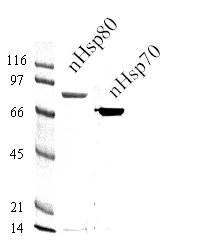
Figure 2. Purification of nHsp70 and nHsp80. Ten-μg samples of nHsp70 and nHsp80 obtained from the final step shown in the Flowchart were subjected to resolution by SDS-PAGE and stained with Coomassie blue. Molecular mass markers (kDa) are shown on the left.
2.1.2 Purification of nHsp80: To purify nHsp80, protein in the 0.6 M NaCl wash fraction is precipitated with ammonium sulfate (80% saturation). The pellet is collected by centrifugation at 12 000 g and re-suspended in Buffer C (20 mM Tris-HCl, pH 7.5) containing 30% (saturation) ammonium sulfate. The protein sample is next loaded onto an Octyl-sepharose column equilibrated against the 30% AS solution. Stepwise elution is carried out by using, successively, buffer containing 25, 20, 15, 10, 5 and 0% saturated AS solution. During this step many of the contaminating proteins are removed and the nHsp80-enriched sample is finally recovered in buffer without AS.
2.1.3 The sample form the above step is concentrated and de-salted using Filtron concentrators (50K cutoff) and applied to an anion-exchange Mono Q (Pharmacia) column—attached to a FPLC unit—equilibrated against buffer C. Elution is conducted using a programmed gradient of 0 to 2 M NaCl. Individual fractions are subjected to SDS-PAGE and those enriched in nHsp80 are pooled and concentrated using 50K cutoff filter unit. In our experience, this step results in recovery of a near-homogeneous preparation of nHsp80 (Fig. 2). Identity of the protein is verified by Western blotting using anti-nHsp80 IgG and by MALDI-TOF MS analysis.
3. Separation of Methionine
synthase and high-molecular-mass peroxidase
3.1 The unbound, flow-through fraction collected from the DEAE-cellulose column (1.2) is further processed through several column chromatography steps to eventually yield purified BrnE (Band running near Hsp Eighty, subsequently identified as the cobalamin-independent methionine synthase) and the high-molecular-mass, heat-induced peroxidase. The protein in the flow-through fraction is collected by precipitation with 80% (saturation) AS.
3.1.2 The pellet from 3.1 is re-suspended gently in Buffer D (20 mM phosphate, pH 7.8), the resulting solution being dialyzed overnight against four changes of the same buffer and applied to a TEAE (Triethylamino ethyl cellulose, Sigma)-cellulose column pre-equilibrated against the same buffer. At this stage methionine synthase is located in the flow-through fraction while peroxidase remains bound to the matrix. The procedure bifurcates in the following steps.
4. Purification of methionine
synthase (BrnE)
4.1 The flow-through fraction from 3.2 is loaded directly on to a 10 mm x 20 cm Cibacron Blue 3GA (Sigma) column in Buffer C. The column is washed extensively with the loading buffer the bound protein being eluted with a linear gradient of 0 to 1.75 M NaCl. Fractions containing BrnE (MetS)—as shown by the SDS-PAGE profile—are pooled and protein therein precipitated with 80% saturation of AS.
4.2 The pellet is re-dissolved in the loading buffer containing 30% AS and subjected to hydrophobic chromatography on Octyl Sepharose CL-4B (Sigma) equilibrated with the same solution. The column is washed with AS (30%)-containing buffer and eluted using 20% AS solution. The total protein is recovered by precipitation by addition of AS to 80% saturation.
4.3 The pellet from 4.2 is dissolved in a small volume of Buffer D, dialyzed overnight against 4 x 1L changes of the same, the sample being concentrated and applied to a Mono Q anion-exchange column – FPLC system, equilibrated against the same buffer. Elution is performed with 0 to 2.0 M NaCl using a programmed gradient. The fractions are tested by SDS-PAGE and those exhibiting a single band corresponding to BrnE are concentrated leading to the recovery of a near homogeneous preparation, identified by MALDI-TOF MS as methionine synthase.
5. Purification of HI-Peroxidase
5.1 The protein bound to the column in 3.1 is eluted with a linear NaCl gradient (0 to 1.5 M) and the resulting fractions are monitored for peroxidase activity (3). Active fractions are pooled and the protein collected by precipitation with AS (80% saturation). The precipitate is dissolved in Buffer C – 30% AS and applied to an Octyl-Sepharose column. Elution is conducted in a stepwise manner using the same buffer—containing progressively decreasing AS saturation— as described in the foregoing. Peroxidase is recovered in 20% AS; the resulting solution is dialyzed extensively against Buffer C and applied to a Cibacron Blue column. Elution is performed using a 0 to 1.5 M NaCl gradient, the protein in active fractions is collected by 80% AS precipitation. The pellet is suspended in Buffer D, dialyzed and fractionated on a Mono Q column with a 0 to 2 M NaCl )programmed) gradient for the final step. The emerging fractions are assayed for peroxidase activity and by SDS-PAGE analysis. Enzymatically active fractions, showing a single band are pooled, desalted/concentrated and stored in Buffer D containing 30% glycerol, at – 80oC.
6. Conclusion
The protocol described in the foregoing results in four high-molecular-mass proteins in a near-homogeneous estate from the same initial material. We have verified the identity of each of these proteins by MALDI-TOF mass spectrometry and by Western blot analysis using specific polyclonal antibodies prepared against them. The final yield for each protein, from a 5-g preparation, is ~3 milligram. The procedure can be completed in 8 to 10 long days and is readily scaled up to generate 8 to 10 mg protein from 15 to 20g of mycelium powder without incurring significant loss in final yield. Only four buffers and five separation matrices are utilized: (i) anion exchangers DEAE-, TEAE-cellulose (or Sephadex), Mono Q; (ii) Octyl Sepharose for hydrophobic chromatography (iii) affinity chromatography Cibacron Blue and ATP agarose (for Hsps) for affinity chromatography. Although the focus of this project was on Neurospora proteins, this approach is applicable to purification of orthologs from other filamentous fungi as stress proteins are known to display a high degree of sequence conservation. Moreover, it can be adapted for similar endeavours to achieve purification of multiple proteins/enzymes in other organisms in a relatively short time. If enzymatic or other facile assays are available, eluates from the columns can be monitored directly with considerable saving of time.

Acknowledgements
The contributions of Carol C. Curle, Derrick Freitag, Patricia Ouimet, Mark Britton and D. Duong during various phases of this project are gratefully acknowledged.
References
Methods in Enzymology (1990) volume 182. A Guide to Protein Purification. Edited by M.P. Deutscher. Academic Press.
Laemmli, U. K. 1970. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature (Lond) 227:680-685.
Hoffman, P.S., Krieg, N.R., and Smibert, R.W. 1979. Studies of microaerophilic nature of Campylobacter fetus subsp. jejuni. I. Physiological aspects of enhanced aerotolerance. Can. J. Microbiol. 25:1-7