Evaluation of automated cell disruptor methods for oomycetous and
ascomycetous model organisms
Takao Kasuga* and Mai Q. N. Bui
USDA-ARS Crop Pathology and Genetics Research Unit, Department of Plant
Pathology, University of California Davis, One Shields Avenue, Davis, CA 95616,
USA (tkasuga@ucdavis.edu)
Fungal Genetics Reports 58:4-
Two automated cell disruptor-based methods for RNA extraction, disruption of
thawed cells submerged in TRIzol Reagent (method QP), and direct disruption of
frozen cells on dry ice (method CP), were optimized for a model oomycete,
Phytophthora capsici, and a model
filamentous ascomycete, Neurospora crassa. The
results were compared with more conventional methods of manual grinding in a
mortar and pestle under liquid nitrogen (method M&P) and those using lyophilized
samples. A chip-based
electrophoresis system showed that methods CP and M&P yielded high integrity RNA
from both P. capsici and
N. crassa.
In contrast, method QP and lyophilized sample-based methods resulted in
inconsistent RNA integrity between the two organisms, indicating they are not
safe alternatives for method M&P.
Microarray mRNA profiling for P. capsici
revealed alterations in global mRNA profiles in those samples that the
chip-based electrophoresis detected slight decreases in RNA integrity.
Despite this, RNA integrity of these samples could still be high enough
to pass conventional stringent quality control measures.
This demonstrated the necessity of global mRNA profiling for the
evaluation of RNA extraction protocols.
Emerging high-throughput transcriptome analysis technologies such as microarray
mRNA profiling (DeRisi et al., 1997)
and more recently RNA-Seq (Nagalakshmi
et al., 2008)
enable us to monitor thousands of gene activities simultaneously and greatly
facilitate the study of functional genomics.
In responding to the ever-growing demand for
transcriptomic analysis for large numbers of samples, automated cell
disruptor-based methods for high-throughput RNA extraction are gaining
popularity
(Van der Vorst et al., 2009).
It should be emphasized that sensitivity and
accuracy of high-throughput transcriptome analysis relies heavily upon the
quality of input RNA samples.
It is not clear if automated cell disruption is
appropriate for high-throughput transcriptome analysis or if it is compatible
with only certain tissue types.
Unlike genomic DNA, mRNA transcripts are extremely sensitive to growth
conditions of the cell. Transcript levels can change on a minute scale due to
both degradation and de novo transcription.
Consequently, alterations in transcript profiles
that are not representative for the condition of interest are easily introduced
during sample harvesting (Fleige
& Pfaffl, 2006; Pieterse
et al., 2006)
and/or during sample preparation.
Accordingly, in order to correctly interpret the
mRNA profiling data, extent of such experimental noise needs to be evaluated and
minimized.
As a means to minimize such unwanted changes, biological
samples are traditionally snap-frozen in liquid nitrogen, and kept at -80°C.
RNA extraction is then carried out by means of a
mortar and pestle with liquid nitrogen (Sambrook
& Russel, 2006).
Ground tissues are kept frozen and subsequently
transferred to a tube containing extraction buffer and are homogenized
immediately.
A chaotropic reagent in the extraction buffer, such as
guanidinium thiocyanate (Chomczynski
& Sacchi, 1987), denatures and deactivates
enzymatic activities, such as nuclease and mRNA transcription activities, thus
protecting RNA integrity.
In contrast, in a typical automated cell disruption
method, beads such as small glass, ceramic or steel beads and extraction buffer
are added to frozen cell cultures resting on an ice bath prior to cell
disruption.
Thus, unlike the traditional mortar and pestle-based
method, samples are inevitably thawed on ice or in the extraction buffer prior
to mechanical disruption of the cell.
Judging from the inherent instability of mRNA
transcripts, thawing can perturb global mRNA profiles, thus introducing
experimental noise.
To our surprise that reproducibility of global mRNA
profiling data for cell disruptor-based methods has not been evaluated.
Furthermore, no standardized protocols are available
for cell disruptor-based methods for fungi or oomycetes.
In this study, a model oomycete
Phytophthora
capsici (Lamour
et al., 2007),
and a model filamentous ascomycete,
Neurospora crassa
(Galagan et
al., 2003),
were chosen as models, and frozen as well as lyophilized samples were subjected
to cell disruption.
There are two main methods for cell disruption; (1)
disruption of cells submerged in extraction buffer, and (2) that under liquid
nitrogen or dry ice without extraction buffer.
Choice of beads for cell disruption is also crucial.
Four distinctive beads with different sizes and
materials; Lysing Matrix A (MP Biomedicals, Solon OH, USA, sold for lysis of
hard samples such as cartilage, bone and seed), Lysing Matrix C (MP Biomedicals,
for lysis of yeast, algae and fungi), Lysing Matrix D (MP Biomedicals, for lysis
of plant and animal tissue) and Lysing Matrix R, which was a home made mixture
of two different sized glass beads, were used in combination with the two
aforementioned cell disruption methods, and evaluated for RNA yield.
RNA integrity and quality was
then evaluated by means of a chip-based electrophoresis system for the two model
organisms, and microarray mRNA profiling for
P. capsici.
We found that global mRNA profiles were perturbed in
response to thawing and lyophilization of samples.
GO ontology enrichment analysis was then used to
infer cellular activities which were responsible for the alteration of mRNA
profiles.
A means to minimize experimental noise associated with cell
disruptor-based protocols are discussed.
As
with method QP, for method CP, four 2 ml screw-cap microcentrifuge tubes, each
containing approximately 100 mg of a frozen mycelium were prepared in
triplicate.
However, for method CP, the microcentrifuge tubes were kept
on a microcentrifuge tube rack (Subzero Blue IsoFreeze Flipper Rack, GeneMate;
BioExpress, Kaysville, UT, USA) on dry ice.
Into each microcentrifuge tube one of each of the
four lysing matrices, chilled at -20°C, was added.
The cells were then disrupted at a speed setting of
6 meters/second for 40 seconds, twice, in a CoolPrep adapter, which was filled
with crushed dry ice.
One ml of TRIzol Reagent was then added to the
pulverized sample, and vigorously vortexed until the sample was completely
thawed and homogenized.
Total RNA was subsequently extracted according to
the manufacturer’s protocol for TRIzol.
In addition, two widely used methods for RNA extraction
from lyophilized samples were adapted for an automated cell disrupter.
Fresh cell cultures or those stored in -80°C,
approximately 100 mg each, were snap-frozen in liquid nitrogen and subjected to
lyophilization for 24 hours under 20 to 100 mTorr.
For each model organism three separate
lyophilizations were conducted for biological controls.
Lyophilized samples were either pulverized with
Lysing Matrices A at room temperature on a QuickPrep adapter (method LQ,
treatment 7 in Table 2) or snap-frozen in liquid nitrogen and pulverized on a
CoolPrep adapter with dry ice (method LC, treatment 6).
For both methods, cells were disrupted at a speed
setting of 6 meters/second for 40 seconds, twice.
One ml of TRIzol Reagent was then added and RNA
extraction was carried out as method CP described above.
Up to 100ug of total RNA was further cleaned using the RNeasy mini protocol for
RNA cleanup (Qiagen, Valencia, CA, USA).
RNA purity and concentration were determined by
NanoDrop 3300 spectrophotometer (Thermo Scientific, Waltham, MA, USA) with
RiboGreen fluorescent dye (Invitrogen Life Technologies).
Integrity of RNA was determined by both agarose gel
electrophoresis and Experion automated electrophoresis system (Bio-Rad,
Hercules, CA, USA).
Experion’s RNA Quality Indicator (RQI), is
independent of sample concentration, and is equivalent to RNA Integrity Number
(RIN, Agilent Technologies).
Although, different algorithms are employed, RQI and
RIN are adjusted to give approximately the same values for identical RNA samples
(Denisov
et al., 2008).
Microarray
design
The
P. capsici whole-genome
expression oligonucleotide 60-mer arrays (4x72K multiplex format, Roche
NimbleGen, Madison, WI, USA) were designed by the manufacturer on the basis of
17,383 gene models derived from the
P. capsici
database at DOE Joint Genome Institute (PhycaF7_best_transcripts.fasta.gz;
http://genome.jgi-psf.org/PhycaF7/PhycaF7.home.html).
On average four probes per open
reading frame (ORF), a total of 69,421 probes for 17,363 gene models, were
synthesized on each of the four arrays on the multiplex array slide.
cDNA
synthesis, hybridization and image acquisition
cDNA synthesis, labeling, hybridization procedure, data acquisition and
normalization were carried out according to the manufacturer’s instructions
(Roche NimbleGen). Briefly,
10 ug of total RNA and oligo dT primer were used to synthesize the first strand
of cDNA, which was followed by the synthesis of the second strand of cDNA to
yield double stranded cDNA.
The Cy3 cyanine dye-labeled random 9-mers were then
used to label the cDNA.
The cDNA was then precipitated with isopropanol,
vacuumed dried, and afterward used for hybridization.
Hybridization was done at 42°C for 16 to 20 hours on
a MAUI Hybridization System (BioMicro Systems, Salt Lake City, UT, USA).
After three steps of washing, microarrays were
scanned on an Axon GenePix 4000B (Molecular Devices, Sunnyvale, CA, USA).
Quantile normalization and background correction
across arrays were performed using Robust Multi-chip Average (RMA) algorithm
(Irizarry et
al., 2003)
implemented in NimbleScan Version 2.5 software.
A MIAME-compliant microarray dataset
(Brazma et al., 2001)
has been deposited in Filamentous Fungal Gene Expression Database at Yale
University (http://bioinfo.townsend.yale.edu/)
(Zhang & Townsend,
2010).
Table S1 lists mRNA profiling results and functional
annotations.
Between hierarchical clusters of cDNA samples, two-sample t-test was used to
identify differentially expressed genes using the software R 2.7.1.
In order to identify difference in cellular
activities between the hierarchical clusters, gene ontology (GO)-based
functional enrichment analysis was conducted.
Gene ontology (GO) annotation
(Harris et al., 2008)
for P. capsici
genome was obtained from the DOE Joint Genome Institute (JGI, PhycaF7_GO.tab.gz;
http://genome.jgi-psf.org/PhycaF7/PhycaF7.download.ftp.html).
Out of the 17,363
P. capsici gene
models that were represented on the microarray, a total of 1,677 GO terms were
assigned to 7,767 gene models according to the annotation scheme conducted by
JGI.
Over- or under- representation of GO terms in
differentially expressed gene groups in relation to the genome was evaluated
against an expected hypergeometric distribution using Fisher’s exact test in the
software R 2.7.1.
A significant level of 0.05 was used with multiple
testing corrections according to Benjamini and Hochberg
(Benjamini & Hochberg, 1995).
Results
Representatives of oomycetes and filamentous ascomycetes,
Phytophthora
capsici and
Neurospora crassa,
respectively, were used for optimization of RNA
isolation using a bench-top cell disruptor, FastPrep-24 Instrument (MP
Biomedicals).
Integrity and global mRNA
profiles of obtained RNA specimens were then cross-examined with total RNA
isolated by mortar and pestle under liquid nitrogen (M&P) and two
lyophilization-based methods.
Optimization
of RNA extraction using a cell disruptor
Cell disruptors are widely used for DNA and RNA extraction as high-throughput
alternatives for mortar and pestle.
Several different methods of cell disruption and
various lysing matrix beads, with which tissues are pulverized, are available.
First, we searched for a method that gives high
quality RNA at high and consistent yields.
Two distinctive cell disruption methods, each with a
specialized adaptor, were tested.
For the first cell disruptor method, 4°C TRIzol
Reagent was added to frozen tissues, and homogenized immediately on a QuickPrep
adaptor (method QP).
For the second method, frozen tissues were
pulverized at a frozen state on a dry ice-filled CoolPrep Cryogenic Adapter, and
subsequently 4°C TRIzol Reagent was added and vortexed (method CP).
For both methods, four distinctive lysing matrix
beads were evaluated for RNA yield: i.e. Lysing Matrix A, Lysing Matrix C,
Lysing Matrix D, and Lysing Matrix R (details in
Introduction and Materials and methods).
Additionally, two lyophilized sample-based methods,
samples disrupted on a dry ice-filled CoolPrep adapter with Lysing Matrix A
(method LC) and samples disrupted on a QuickPrep adapter at room temperature
with Lysing Matrix A without extraction buffer (method LQ), were conducted.
Similar trends for performance of
extraction methods for P. capsici and
N. crassa
were recognized.
For method QP, the yield difference due to lysing
matrix beads were small (Table 1).
However, for
N. crassa Matrix R
significantly underperformed any other lysing matrices (p<0.001).
For method CP, difference in performance of matrices
was more prominent than that of method QP.
Matrices A and R, which were
comprised of fine and large beads, outperformed Matrices C and D, both of which
were comprised of small uniform beads, for
P. capsici
(t test, p<0.01) and for
N. crassa (p<0.001).
RNA yields from lyophilized samples (methods LC and
LQ) and those from a mortar and pestle under liquid nitrogen (method M&P) were
comparable to the highest values for method CP.
The quality of RNA obtained by the five methods, QP,
CP, LC, LQ and M&P, judged by intensity of 28S and 18S ribosomal RNA bands on
agarose gel, was indistinguishable (data not shown).
|
Table 1. Summary for total RNA yield due to
different methods |
|||||
|
Methoda |
Lysing matrixb |
No. replicates |
Average weight mg |
Average total RNA yield µg |
Yield (1SD) µg/mg-tissue |
|
P. capsici |
|
|
|
|
|
|
QP |
A |
3 |
96.7 |
78.6 |
0.8 (0.5) |
|
QP |
C |
3 |
86.7 |
87.3 |
1.0 (0.8) |
|
QP |
D |
3 |
86.7 |
100.6 |
1.2 (0.2) |
|
QP |
R |
3 |
100.0 |
82.0 |
0.8 (0.4) |
|
CP |
A |
3 |
86.7 |
129.9 |
1.5 (0.4) |
|
CP |
C |
3 |
93.3 |
14.1 |
0.2 (0.4) |
|
CP |
D |
3 |
80.0 |
66.3 |
0.8 (0.6) |
|
CP |
R |
3 |
83.3 |
119.3 |
1.4 (0.6) |
|
LC |
A |
3 |
78.3 |
133.4 |
1.6 (0.6) |
|
LQ |
A |
3 |
77.0 |
108.0 |
1.4 (0.2) |
|
M&P |
|
1 |
225.0 |
343.5 |
1.5 (0.0) |
|
|
|
|
|
|
|
|
N. crassa |
|
|
|
|
|
|
QP |
A |
3 |
97.0 |
751.5 |
7.7 (1.0) |
|
QP |
C |
3 |
98.3 |
513.1 |
5.2 (0.5) |
|
QP |
D |
3 |
100.7 |
575.3 |
5.7 (0.3) |
|
QP |
R |
3 |
100.3 |
302.2 |
3.0 (0.3) |
|
CP |
A |
3 |
103.3 |
601.6 |
5.8 (0.9) |
|
CP |
C |
3 |
99.0 |
94.3 |
1.0 (0.2) |
|
CP |
D |
3 |
102.3 |
28.3 |
0.3 (0.4) |
|
CP |
R |
3 |
106.7 |
902.0 |
8.5 (1.4) |
|
LC |
A |
3 |
94.7 |
796.4 |
8.4 (1.1) |
|
LQ |
A |
3 |
103.3 |
670.5 |
6.5 (2.3) |
|
M&P |
|
1 |
210.0 |
1326.3 |
6.3 (0.0) |
|
a
QP: samples submerged in TRIzol, homogenized on
a QuickPrep adapter. |
|||||
|
CP: frozen samples disrupted on a dry ice-filled
CoolPrep adapter. |
|||||
|
LC: lyophilized sample disrupted on a dry
ice-filled CoolPrep adapter. |
|||||
|
LQ: lyophilized samples disrupted on a QuickPrep
adapter at room temperature. |
|||||
|
M&P: a mortar and pestle under liquid nitrogen. |
|||||
|
b
various beads for cell disruption.
Details in Introduction. |
|||||
Experimental design for evaluation of cell disruptor protocols by means of a
chip-based capillary electrophoresis and microarray mRNA profiling
With appropriate combinations of cell lysing matrices and adaptors, the high
throughput cell disruptor was shown to yield a comparable quantity of RNA in
comparison to method M&P.
The next objective was to examine whether RNA
samples derived from the cell disruptor, fresh or lyophilized samples, had high
integrity and were able to reproduce global mRNA profiles of RNA samples derived
from method M&P.
Especially, unlike M&P, in method QP, TRIzol Reagent
is needed to add to samples prior to cell disruption, which could potentially
impact global mRNA profiles.
Also, effect of lyophilization on transcriptome has
not been investigated.
For each of the five RNA isolation methods, the number of biological replicates
used for the measurement of RQI values and sample names of biological replicates
used for microarray profiling were shown in Table 2.
For the QP method, no more than four samples were
handled simultaneously, and no longer than 2 minutes had elapsed between
unscrewing caps for the addition of TRIzol Reagent to frozen samples and the
initiation of mechanical disruption.
|
Table 2. Effects of post-harvest treatments on
the integrity of RNA samples |
||||||
|
Treatment No. |
Post-harvest treatmenta |
Homogenization |
Lysing Matrix |
n |
Average RQI |
Samples used for microarray |
|
P. capsici |
|
|
|
|
|
|
|
|
no |
M&P |
- |
3 |
9.8 |
MP1 |
|
|
no |
CP |
A |
5 |
9.5 |
CP1, CP2, CP3 |
|
|
no |
QP |
R |
2 |
9.5 |
QP1, QP2b, QP3b |
|
|
|
|
|
|
|
|
|
1 |
4oC
TRIzol 10min |
QP |
R |
3 |
9.6 |
TR1, TR2 |
|
2 |
4oC
10min |
QP |
R |
3 |
9.3 |
FT2, FT3 |
|
3 |
RNALater ICE, -20oC, 1 day |
QP |
R |
3 |
- |
|
|
4 |
RNALater, 4oC, 1 day |
QP |
R |
1 |
9.7 |
RL2 |
|
5 |
Never frozen, 4C 10min |
QP |
R |
2 |
9.5 |
NF1 |
|
6 |
lyophilization |
CP (LC) |
A |
6 |
8.1 |
LC1, LC2 |
|
7 |
lyophilization |
QP w/o buffer (LQ) |
A |
6 |
6.8 |
LQ1, LQ2 |
|
|
|
|
|
|
|
|
|
N. crassa |
|
|
|
|
|
|
|
|
no |
M&P |
- |
1 |
9.7 |
|
|
|
no |
CP |
A |
1 |
9.7 |
|
|
|
no |
QP |
R |
4 |
9.4 |
|
|
|
|
|
|
|
|
|
|
1 |
4oC
TRIzol 10min |
QP |
R |
5 |
8.9 |
|
|
2 |
4oC
10min |
QP |
R |
2 |
9.2 |
|
|
3 |
RNALater ICE, -20oC, 1 day |
QP |
R |
2 |
- |
|
|
4 |
RNALater, 4oC, 1 day |
QP |
R |
2 |
- |
|
|
6 |
lyophilization |
CP (LC) |
A |
3 |
9.2 |
|
|
7 |
lyophilization |
QP w/o buffer (LQ) |
A |
3 |
8.8 |
|
|
a
All samples except for treatment 5 were
snap-frozen in liquid nitrogen prior to
post-harvest treatment. |
||||||
|
b
QP2 and QP3 are technical replicates; cDNAs were
prepared from the same RNA sample with separate
reverse transcription reactions. |
||||||
Additionally, we simulated a situation where a large number of samples are
handled together.
Commercially available cell disruptor such as Mini-Beadbeater
(Biospec, Bartlesville, OK) and Precellys Homogenizer (Bertin Technologies,
Montigny-le-Bretonneux, France) are able to hold 45 and 24 microcentrifuge
tubes, respectively.
The maximum sample load for a QuickPrep adaptor on
FastPrep 24 is 24 microcentrifuge tubes.
When 24 samples are handled, 5 to 10 minutes is
needed between unscrewing the first sample and initiation of a homogenizer.
In such a scenario, if samples are kept in an ice
bath, they are exposed to 0°C or a higher temperature, and thus there is an
opportunity for thawing.
Upon the addition of 4°C TRIzol Reagent, samples
will inevitably be thawed in a short period of time.
It is possible that prolonged duration in a thawed
state could introduce noise to mRNA profiles.
In order to account for high-throughput RNA
extraction, two additional treatments were designed for method QP (Table 2).
First, approximately 100 mg
each of frozen
P. capsici tissues were
incubated with 4°C TRIzol Reagent in a 4°C water bath for 10 minutes to evaluate
the effects of TRIzol and thawing (Table 2, treatment 1).
Frozen samples of
P. capsici were
also incubated in microcentrifuge tubes in a 4°C water bath to assess the
thawing effect alone (treatment 2).
In addition, RNAlater (Ambion), an aqueous reagent
widely used to preserve RNA in fresh animal tissues (treatment 4)
(Florell
et al., 2001; Mutter
et al., 2004),
and RNALater-ICE (treatment 3) used to preserve RNA in previously frozen animal
tissues
(Li
et al., 2004),
were tested for the hope that they were capable of preserving mRNA profiles in
P. capsici
after harvest.
In order to assess mRNA response to cold-shock
alone, cultures were harvested and transferred to microcentrifuge tubes, without
liquid nitrogen snap-freeze, and incubated at 4°C for 10 minutes (treatment 5).
The two lyophilized sample-based methods (treatments
6 and 7), were also included for evaluation of their mRNA quality.
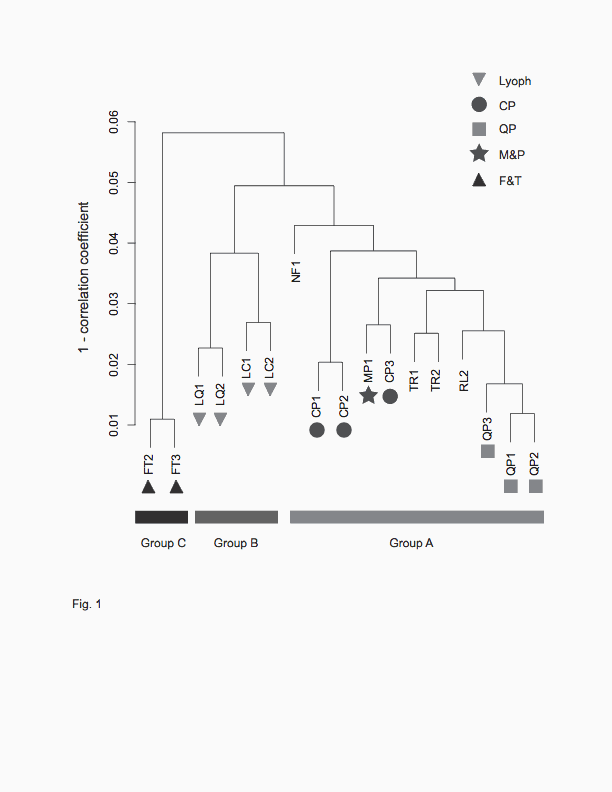
Fig. 1. Hierarchical clustering of cDNA samples with various post-harvest
treatments. 17 samples were clustered based on their expression patterns of
12,504 genes. The seven samples used for the optimization of cell disruption
methods, CP, QP and M&P were indicated with filled circle, filled square and
filled star, respectively. Group B is comprised of lyophilized samples
(indicated with filled inverted triangles), and group C is comprised of samples
which were thawed and incubated without TRIzol Reagent (indicated with filled
triangles). All the remaining samples were found in group A.
|
Table
3. Enriched GO biological processes in response
to lyophilization and freeze and thaw (complete
list in Table S2) |
|||||
|
GO |
descriptiona |
Obsb |
Expb |
Totalc |
P
valued |
|
|
A>B |
|
|
|
|
|
GO:0006007 |
glucose catabolic process |
15 |
7.1 |
41 |
2.6E-02 |
|
GO:0046164 |
alcohol catabolic process |
15 |
7.7 |
44 |
4.5E-02 |
|
|
|
|
|
|
|
|
|
B>A |
|
|
|
|
|
GO:0016310 |
phosphorylation |
105 |
63.7 |
399 |
2.1E-06 |
|
GO:0007017 |
microtubule-based process |
33 |
13.1 |
82 |
2.6E-06 |
|
GO:0007166 |
cell surface receptor linked signal transduction |
10 |
4.0 |
25 |
2.7E-02 |
|
|
|
|
|
|
|
|
|
A>C |
|
|
|
|
|
GO:0006519 |
cellular amino acid and derivative metabolic
process |
65 |
41.4 |
186 |
2.2E-03 |
|
|
|
|
|
|
|
|
|
C>A |
|
|
|
|
|
GO:0007017 |
microtubule-based process |
25 |
12.5 |
82 |
7.3E-03 |
|
|
|
|
|
|
|
|
|
C>B |
|
|
|
|
|
GO:0034961 |
cellular biopolymer biosynthetic process |
70 |
33.2 |
355 |
4.5E-08 |
|
GO:0010467 |
gene expression |
69 |
35.6 |
381 |
2.1E-06 |
|
a
Enriched GO terms between hierarchical clusters
defined in Fig. 1. |
|||||
|
b
Observed number and expected number of genes if
probabilities of each outcome are independent of
the cluster. |
|||||
|
c
Total number of detected genes on the
microarray. |
|||||
|
d
P values were determined using Fisher's
exact test with Benjamini and Hochberg multiple
testing correction. |
|||||
There is a growing demand for high-throughput RNA isolation, and automated cell
disruptors have been gaining popularity
(Van der Vorst
et al., 2009).
Validation of the technology is, however, limited to
1) RNA integrity measured by capillary electrophoresis and 2) mRNA profiles of a
handful of genes measured by real time quantitative PCR
(Van
der Vorst et
al., 2009).
In this research we first
optimized automated cell disruptor protocols for an oomycete plant pathogen
P. capsici
and a filamentous ascomycete
N. crassa and then
inspected RNA quality by capillary electrophoresis for both
N. crassa
and P. capsici and by
global mRNA profiling for only
P. capsici.
We found that lyophilization as well as thawing of samples prior to the addition
of extraction buffer resulted in significant alteration of mRNA profiles.
This finding is notable becase thawing is inevitable
for the majority of protocols devised with cell disruptors, (e. g.
(Kasuga & Glass, 2008; Monteiro
et al., 2009; Rautio, 2010))
as well as the hot phenol protocol for frozen yeast cells (e.g.
(Castillo et
al., 2002; Eelderink-Chen
et al., 2010)).
It should be emphasized that the effect of thawing,
which is evident from microarray mRNA profiling, cannot be explicitly detected
by conventional quality control measures.
For method QP for
P. capsici,
it was found that adding TRIzol Reagent to frozen samples could minimize the
change of mRNA profiles due to thawing.
This indicates that TRIzol
Reagent percolates into P. capsici cells
and ceases enzymatic activity; as a consequence this prevents the alteration of
mRNA profiles.
In contrast, a 4°C TRIzol
treatment was found to compromise RNA integrity in
N. crassa.
This implies that the cell wall
of N. crassa
is less permeable to TRIzol, allowing RNA to degrade.
We do not have mRNA profiling
data for N.
crassa, however, it is likely that addition of
TRIzol also affects its transcriptome.
We, therefore do not recommend the use of method QP
unless effects of TRIzol on the preservation of mRNA of particular species or
tissue types being investigated has properly been evaluated by means of global
mRNA profiling.
RNAlater (Ambion), is an aqueous reagent, and is widely
used to stabilize and protect RNA in animal tissue
(Mutter et al., 2004).
The global mRNA profile of the
RNAlater-treated
P. capsici sample closely
resembles that prepared by M&P, indicating that the reagent immediately
permeated the tissue and stabilized mRNA.
On the other hand, an RNALater
treatment resulted in severely degraded RNA from
N. crassa.
This is consistent with our
finding that while a 4°C TRIzol treatment successfully preserved RNA integrity
in P. capsici,
the same treatment on
N. crassa yielded slightly
degraded RNA.
We again do not recommend RNALater unless its
effectiveness has been proved for interrogated species or tissue types.
Lyophilization is another widely used method to cease
cellular activity prior to RNA extraction
(Leary
et al., 1969; Sanchez-Rodriguez
et al., 2008).
Lyophilized samples can be effectively pulverized on
a mortar and pestle or an automated cell disruptor.
We found that lyophilization
damaged RNA integrity and altered mRNA profiles in
P. capsici.
The adverse effect of
lyophilization was also observed for
N. crassa
but to a lesser extent.
In lyophilization, samples frozen in liquid nitrogen
are placed in a vacuum chamber at room temperature, where gas pressure is
typically at 100 mTorr.
Because the chamber pressure is below the saturation
pressure of water vapor, sublimation will progress, while removal of heat by
sublimation keeps the sample frozen.
It is possible that a rigid cell wall and increasing
concentrations of solutes in the cytosol effectively prevent the cell from
complete desiccation, by which the cell maintains residual cellular activity
such as de novo transcription and mRNA degradation.
GO enrichment analysis revealed that in the
lyophilized samples 105 genes with a GO term “phosphorylation”, of which 53 were
annotated as “kinases”, and also 41 genes with a GO term “cell communication”
appeared to be increased.
It seems that
P. capsici is
capable of activating signal transduction cascades in response to
lyophilization.
Reactivation of transcriptional machinery likely
happens when samples in the vacuum chamber are thawed during lyophilization.
Again, the effect of lyophilization, which is
evident from microarray mRNA profiling, cannot be detected by conventional
quality control measures with certainty.
Thawed samples also showed a large alteration in the
transcriptome.
A cryoinjury of the cell is likely responsible for the
shift of mRNA profiles.
During freezing, formation of intracellular ice and
shrinkage of cells occur in various ascomycete and oomycete species
(Morris
et al., 1988),
which lead to loss of integrity of cell membrane, organelles, and thus
viability.
GO enrichment analysis showed that in response to freeze
and thaw, genes for microtubule motors were activated.
Lyophilized samples, which were also likely to have
incurred cryoinjury, showed activation of genes for microtubule motors.
Taken together, activation of microtubule motor
might be associated with cell repair.
In summary, we demonstrated that a transcriptome is
extremely sensitive to RNA extraction protocols.
A brief thawing of samples on ice or in contact with
extraction buffer or lyophilization process can significantly alter mRNA
profiles.
We have analyzed mRNA profiles for
only the oomycete
P. capsici, however, such
alteration in transcriptomes is likely to occur for the ascomycete
N. crassa as well
as diverse organisms e.g. bacteria, plants and animals.
We recommend the use of a mortar and pestle or
frozen-phase cell-disruption method unless application of RNAlater or TRIzol-submerged
cell disruption method is proven safe by means of global mRNA profiling.
Benjamini, Y., Y. Hochberg, 1995. Controlling the
false discovery rate - a practical and powerful approach to multiple testing.
Journal of the Royal Statistical Society
Series B-Methodological 57: 289-300.
Brazma, A., P. Hingamp, J. Quackenbush & other
authors, 2001. Minimum information about a microarray experiment (MIAME) -
toward standards for microarray data. Nat Genet 29: 365-371.
Castillo, E. A., J. Ayte, C. Chiva, A. Moldon, M.
Carrascal, J. Abian, N. Jones, E. Hidalgo, 2002. Diethylmaleate activates the
transcription factor Pap1 by covalent modification of critical cysteine
residues. Mol Microbiol 45: 243-254.
Chomczynski, P., N. Sacchi, 1987. Single-step
method of RNA isolation by acid guanidinium thiocyanate phenol chloroform
extraction. Anal Biochem 162: 156-159.
Copois, V., F. Bibeau, C. Bascoul-Mollevi & other
authors, 2007. Impact of RNA degradation on gene expression profiles: Assessment
of different methods to reliably determine RNA quality.
J Biotechnol 127: 549-559.
Denisov, V., W. Strong, M. Walder, J. Gingrich,
H. Wintz, 2008. Development and validation of RQI: an RNA quality indicator for
the Experion automated electrophoresis system.
Bio-Rad Bulletin #5761.
DeRisi, J., V. R. Iyer, P. O. Brown, 1997.
Exploring the metabolic and genetic control of gene expression on a genomic
scale. Science 278: 680-686.
Dudoit, S., Y. H. Yang, M. J. Callow, T. P.
Speed, 2002. Statistical methods for identifying differentially expressed genes
in replicated cDNA microarray experiments.
Statistica Sinica 12: 111-139.
Eelderink-Chen, Z., G. Mazzotta, M. Sturre, J.
Bosman, T. Roenneberg, M. Merrow, 2010. A circadian clock in
Saccharomyces cerevisiae.
Proc Natl Acad Sci U S A 107:
2043-2047.
Englander, L., L. F. Roth, 1980. Interaction of
light and sterol on sporangium and chlamydospore production by
Phytophthora lateralis.
Phytopathology 70: 650-654.
Fleige, S., M. W. Pfaffl, 2006. RNA integrity and
the effect on the real-time qRT-PCR performance.
Mol Asp Med 27: 126-139.
Fleige, S., V. Walf, S. Huch, C. Prgomet, J. Sehm,
M. W. Pfaffl, 2006. Comparison of relative mRNA quantification models and the
impact of RNA integrity in quantitative real-time RT-PCR.
Biotechnol Lett 28: 1601-1613.
Florell, S. R., C. M. Coffin, J. A. Holden, J. W.
Zimmermann, J. W. Gerwels, B. K. Summers, D. A. Jones, S. A. Leachman, 2001.
Preservation of RNA for functional genomic studies: A multidisciplinary tumor
bank protocol. Mod Pathol 14: 116-128.
Galagan, J. E., S. E. Calvo, K. A. Borkovich &
other authors, 2003. The genome sequence of the filamentous fungus
Neurospora crassa.
Nature 422: 859-868.
Harris, M. A., J. I. Deegan, J. Lomax & other
authors, 2008. The Gene Ontology project in 2008.
Nucleic Acids Res 36: D440-D444.
Irizarry, R. A., B. Hobbs, F. Collin, Y. D.
Beazer-Barclay, K. J. Antonellis, U. Scherf, T. P. Speed, 2003. Exploration,
normalization, and summaries of high density oligonucleotide array probe level
data. Biostatistics 4: 249-264.
Kasuga, T., N. L. Glass, 2008. Dissecting colony
development of Neurospora crassa using
mRNA profiling and comparative genomics approaches.
Eukaryot Cell 7: 1549-1564.
Lamour, K. H., J. Win, S. Kamoun, 2007. Oomycete
genomics: new insights and future directions.
FEMS Microbiol Lett 274: 1-8.
Leary, J. V., A. J. Morris, A. H. Ellingbo, 1969.
Isolation of functional ribosomes and polysomes from lyophilized fungi.
Biochim Biophys Acta 182: 113-120.
Li, M., W. Li, H. J. Min, Q. Z. Yao, C. Y. Chen,
W. E. Fisher, 2004. Characterization of somatostatin receptor expression in
human pancreatic cancer using real-time RT-PCR.
J Surg Res 119: 130-137.
Monteiro, J. P., K. V. Clemons, L. F. Mirels, J.
A. Coller, T. D. Wu, J. Shankar, C. R. Lopes, D. A. Stevens, 2009. Genomic DNA
microarray comparison of gene expression patterns in
Paracoccidioides brasiliensis mycelia
and yeasts in vitro. Microbiology-Sgm
155: 2795-2808.
Morris, G. J., D. Smith, G. E. Coulson, 1988. A
comparative-study of the changes in the morphology of hyphae during freezing and
viability upon thawing for 19 Species of fungi.
Cryobiology 25: 517-517.
Mutter, G. L., D. Zahrieh, C. M. Liu, D. Neuberg,
D. Finkelstein, H. E. Baker, J. A. Warrington, 2004. Comparison of frozen and
RNALater solid tissue storage methods for use in RNA expression microarrays.
BMC Genomics 5: 88.
Nagalakshmi, U., Z. Wang, K. Waern, C. Shou, D.
Raha, M. Gerstein, M. Snyder, 2008. The transcriptional landscape of the yeast
genome defined by RNA sequencing. Science 320: 1344-1349.
Pieterse, B., R. H. Jellema, M. J. van der Werf,
2006. Quenching of microbial samples for increased reliability of microarray
data. J Microbiol Methods 64: 207-216.
Rautio, J. J., 2010. Multiplex gene expression
analysis by TRAC in fungal cultures.
Methods Mol Biol 638: 165-173.
Sambrook, J., D. W. Russel, 2006.
The Condensed Protocols from Molecular Cloning: A Laboratory Manual. New
York: Cold Spring Harbor Laboratory Press.
Sanchez-Rodriguez, A., O. Portal, L. E. Rojas, B.
Ocana, M. Mendoza, M. Acosta, E. Jimenez, M. Hofte, 2008. An efficient method
for the extraction of high-quality fungal total RNA to study the
Mycosphaerella fijiensis-Musa spp.
Interaction. Mol Biotechnol 40:
299-305.
Subramanian, A., P. Tamayo, V. K. Mootha & other
authors, 2005. Gene set enrichment analysis: A knowledge-based approach for
interpreting genome-wide expression profiles.
Proc Natl Acad Sci U S A 102:
15545-15550.
Van der Vorst, S., A. F. Dekairelle, L. Irenge,
M. Hamoir, A. Robert, J. L. Gala, 2009. Automated cell disruption is a reliable
and effective method of isolating RNA from fresh snap-frozen normal and
malignant oral mucosa samples. Clin Chem Lab Med 47: 294-301.
Vogel, H. J., 1956. A convenient growth medium
for Neurospora (Medium N).
Microbial Genet Bull 13: 42-43.
Zhang, Z., J. P. Townsend, 2010. The filamentous
fungal gene expression database (FFGED).
Fungal Genet Biol 47: 199-204.