Strategies for the
molecular genetic manipulation and visualization of the human fungal pathogen
Penicillium marneffei
Kylie J. Boyce#, Hayley, E. Bugeja#, Harshini Weerasinghe, Michael J. Payne, Lena Schreider, Changwon Park, Trent Woodward and Alex Andrianopoulos
Department of Genetics, The University of Melbourne,
Parkville, Victoria, Australia.
#
These authors have contributed equally
(pdf)
P. marneffei has
been established as an experimentally amenable system to study morphogenesis and
pathogenicity. This paper describes the development of a number of tools,
including numerous selectable markers, to expand the ease with which it can be
genetically manipulated. Combined with strains engineered for homologous
recombination of exogenous DNA, these tools facilitate efficient molecular
genetic studies.
Introduction
To facilitate molecular genetic analysis of gene function
in P. marneffei, an important opportunistic pathogen of humans, an
efficient DNA-mediated transformation protocol was developed using exogenous DNA
and polyethylene glycol-mediated protoplast fusion
(Borneman et al. 2001).
Spontaneous mutants were also derived from the
P. marneffei type strain FRR2161 (ATCC18224) by selection on the
toxic compounds 5-fluoroorotic acid (5-FOA) and chlorate to generate
pyrG (orotidine 5’-monophosphate
decarboxylase) auxotrophic and niaD
(nitrate reductase) utilization mutants, respectively (Table 1 and 2)(Borneman
et al. 2001). Combined with the
dominant selectable marker of bleomycin/phleomycin resistance and the ability to
recycle the pyrG marker
(Borneman et al. 2001),
complex genetically modified strains can be created for analysis. Exogenous DNA
introduced during transformation preferentially integrates into the genome of
P. marneffei by non-homologous
integration, however, strains defective in the non-homologous end-joining
machinery have recently been developed that result in highly efficient
homologous integration (Table 1)(Bugeja
et al, 2012). This study
describes the development of additional auxotrophic and dominant selectable
markers to broaden the options for selection of transformants containing
introduced DNA in the type strain of P.
marneffei or into clinical isolates (Table 2). In addition, a number of
constructs have been developed for targeted integration at specific loci and for
the rapid generation of gene deletion constructs using the previously described
GatewayTM cloning system (Bugeja
et al, 2012). Lastly, tools for
the microscopic visualization of P.
marneffei mutants generated by these techniques are described.
|
|
||
|
Strain |
Genotype |
Origin |
|
FRR2161 |
Wild type |
Bamboo rat (Rhizomys
sinensis) isolate (Supplied by J. Pitt,
CSIRO Food Industries, Sydney, Australia). Also
available from the American Type Culture
Collection as ATCC18224 |
|
G146 (SPM3) |
niaD1 |
(Borneman
et al. 2001) |
|
G147 (SPM4) |
niaD1 pyrG1 |
(Borneman
et al. 2001) |
|
G809 |
niaD1 pyrG1 ∆ligD::AnpyrG+ |
(Bugeja et
al, 2012) |
|
G816 |
niaD1 pyrG1 ∆ligD |
(Bugeja et
al, 2012) |
|
G779 |
niaD1 pyrG1 ∆riboB::AnpyrG |
This study |
|
G829 |
niaD1 pyrG1 ∆ligD
riboB::AnpyrG |
This study |
|
G780 |
niaD1 pyrG1 ∆riboB |
This study |
|
G890 |
niaD1 pyrG1 ∆ligD
riboB |
This study |
|
G830 |
niaD1 pyrG1 ∆ligD
pyroA::AnpyrG |
This study |
|
G908 |
niaD1 pyrG1 ∆ligD
pyroA |
This study |
|
G487 |
niaD1 pyrG1 areA∆DBD |
This study |
|
G831 |
niaD1 pyrG+
HI::mCherry ∆ligD |
This study |
In order to develop additional selectable markers for use
with auxotrophic P. marneffei strains,
the riboB and
pyroA genes encoding a GTP cyclohydrolase and 5’-phosphate synthase
required for riboflavin and pyridoxine biosynthesis, respectively, were
cloned and deleted (Oakley et al. 1987b; Osmani
et al. 1999). The
P. marneffei riboB gene was PCR
amplified (primers AA18 and AA19) and cloned into pBluescript II SK+
(Stratagene)(pAA7329, Table 3 and 4). A split marker construct was generated by
overlap PCR to facilitate deletion of the
riboB locus using the A. nidulans pyrG
blaster cassette (pHB7131 and pHB7132; Tables 3 and 4). A
riboB gene deletion strain, in which the entire coding region has
been replaced with the A. nidulans pyrG
blaster cassette, was been generated in the SPM4 (G779) and ∆ligD
pyrG- (G829) strains (Table
1). pyrG- derivatives
(G780 and G890) have also been isolated as 5-FOA resistant sectors which have
lost the A. nidulans pyrG gene
(Borneman
et al. 2001). P. marneffei
∆riboB strains require supplementation
with 5 µg mL-1 riboflavin and must be grown in the dark to prevent
photolytic breakdown of riboflavin (Table 2).
|
Table 2.
P.
marneffei selectable marker systems |
|||
|
Selectable marker |
Recipient strain |
Phenotype of
recipient strain |
Selection regime |
|
pyrG |
pyrG1 |
Uracil auxotroph
(requires 10 mM
uracil),
5’FOA resistant |
Positive: No
added uracil
Negative: 1 mg mL-1
5-FOA + 10 mM uracil |
|
niaD |
niaD1 |
Nitrate
utilization defect,
Chlorate
resistant |
Positive: 10 mM
nitrate as the sole nitrogen source
Negative: 10 mM
chlorate |
|
riboB |
DriboB |
Riboflavin
auxotroph (requires 5 µg mL-1
riboflavin & growth in dark) |
No added
riboflavin |
|
pyroA |
DpyroA |
Pyridoxine
auxotroph (requires 1 µg mL-1
pyridoxine) |
No added
pyridoxine |
|
barA |
Any |
N/A |
25-50 µg mL-1
of PPT |
|
ptrA |
Any |
N/A |
0.1-0.2 mg mL-1
of pyrithiamine hydrobromide |
|
barA/hv-tk |
Any |
N/A |
Positive: 25-50
µg mL-1 of PPT
Negative: 5 µM
FDU |
|
areA |
areA∆DBD |
Reduced growth on
nitrogen sources other than ammonium |
10 mM nitrite as
the sole nitrogen source |
|
wA |
Any |
Green conidiation |
Screen for white
conidiation phenotype |
The P. marneffei
pyroA gene was amplified (primers AA20 and AA21), cloned (pAA7331) and a
split marker gene deletion construct was generated using overlap PCR (pHB7129
and pHB7130; Table 3 and 4). A pyroA
gene deletion strain, in which the entire coding region has been replaced with
the A. nidulans pyrG blaster cassette, was generated in the ∆ligD
pyrG- strain (G830) and a
pyrG- derivative (G908) has
also been obtained as described above (Borneman et al. 2001).
Similar to A. nidulans, the
P. marneffei ∆pyroA strain requires supplementation with 1µg mL-1
pyridoxine (Table 2). Despite supplementation, it has been observed that this
strain grows slower than pyroA+
strains and has slightly reduced conidiation at 25°C.
Since it was first developed as a dominant selectable
marker in N. crassa, L-phosphinothricin (PPT), also known as glufosinate
ammonium, resistance has been used in many fungi
(Ahuja and Punekar 2008; Avalos et al.
1989). L-phosphinothricin
inhibits glutamine synthetase, required for ammonium assimilation, by occupying
the substrate (glutamate) pocket (Gill and
Eisenberg 2001). The Aspergillus
oryzae barA gene encodes PPT
resistance and can be used in P. marneffei
for transformation (Table 5). Similar to
A. nidulans and N. crassa,
selection of P. marneffei PPT
resistant transformants requires approximately 25-50 µg mL-1 of PPT
(Table 2)(Ahuja and Punekar 2008).
Table 3. Oligonucleotides used in this
study
|
Name |
Template |
Sequence 5’ – 3’ |
|
AA18 |
riboB |
CAGCGTGTCGAGTTCAGG |
|
AA19 |
riboB |
TCTCTCTCCATCAGCGGC |
|
CC63 |
riboB |
AAGGGTGAACACTATCCTTGCGGAGCGATGT |
|
CC64 |
riboB |
CCGTACATCGCTCCGCAAGGATAGTGTTCAC |
|
CC65 |
riboB |
GATGAGTGGCAGGGGGCTGCAGAAAGGTATA |
|
CC66 |
riboB |
CTCCTTATACCTTTCTGCAGCCCCCTGCCAC |
|
QQ11 |
riboB |
TGACCATGATTACGCCAAGC
|
|
QQ12 |
riboB |
TGAATTCGAGCTCGGTACCC |
|
AA20 |
pyroA |
TCAAGCCGTTACTGTCCA |
|
AA21 |
pyroA |
GACTTTGAACGGCACATT |
|
CC59 |
pyroA |
AAGGGTGAACACTATCCATTCTCGGATGCCATTGC |
|
CC60 |
pyroA |
GCAATGGCATCCGAGAATGGATAGTGTTCACCCTT |
|
CC61 |
pyroA |
GATGAGTGGCAGGGGGCTATTGGCGTTAAGCGGTGG |
|
CC62 |
pyroA |
CCACCGCTTAACGCCAATAGCCCCCTGCCACTCATC |
|
NN61 |
ptrA |
TTACGGGATCCCATTGGTAA |
|
NN62 |
ptrA |
AAGACAAATCCGGTTCATGC |
|
AA52 |
pyrG |
CTTATCGGGCCGGAGCA |
|
AA53 |
pyrG |
ATCCTCGCTCTCCTCTTTCT |
|
HH81 |
pyrG |
GAGTTTGTCGTGCTATCCG |
|
II01 |
pyrG |
GTCTTGCTTACGGTACTCC |
|
QQ39 |
pyrG |
CGCAAACTATTCGCTACTG |
|
KK77 |
niaD |
CGAAACCAAGACGAGAGGC |
|
KK78 |
niaD |
TTGGATGGGAAGAGAGCG |
|
II08 |
wA |
AATCTAGAGCCTATGCCTATGTCTTC |
|
II09 |
wA |
AAGCGGCCGCGAATGCGACAGGCGTATC |
|
HH45 |
wA |
GTCATGCAGGAAAGGGTCAT |
|
HH46 |
wA |
GCACCGGTCGATACTTGAAT |
|
LL9 |
mCherry |
ACTAGTGCCGGTGCGCCCGGG |
|
LL10 |
mCherry |
TCTAGACCAGCATCTGATGTCCTGGT |
|
LL11 |
H1 |
GGATCCCCCTTGACAAAACCGCAGAG |
|
LL12 |
H1 |
ACTAGTGGCCTTCTTGGGTGTGGCCT |
|
MM8 |
pDONRTM221-attP2 |
TGAGGACAATAGCGAGTAGG
|
|
MM9 |
pDONRTM221-attP1 |
TGAGGACAATAGCGAGTAGG |
Table 4. Plasmids used in this study
|
Plasmid |
Name |
Description |
|
pPL3 |
An
riboB |
A. nidulans riboB gene (Oakley
et al. 1987b) |
|
pAA7329 |
riboB |
riboB
gene amplified using AA18 and AA19 and cloned
into the
EcoRV site of pBluescript SK+ |
|
pHB7131 |
5’∆riboB::pyrG |
5’
riboB fused to the 5’ half of
pyrG.
Overlap PCR using AA18, CC63, CC64 and AA52.
Amplify using AA18 and AA52. |
|
pHB7132 |
3’∆riboB::pyrG |
3’
riboB fused to the 3’ half of
pyrG.
Overlap PCR using CC65, AA19, CC66 and AA53.
Amplify using AA19 and AA53. |
|
pAA7331 |
pyroA |
pyroA
gene amplified using AA20 and AA21 and cloned
into the
EcoRV site of pBluescript II SK+
(Osmani
et al.
1999). |
|
pHB7129 |
5’∆pyroA::pyrG |
5’
pyrA fused to the 5’ half of
pyrG. Overlap PCR using AA21, CC59, CC60 and
AA52. Amplify using AA21 and AA52. |
|
pHB7130 |
3’∆pyroA::pyrG |
3’
pyroA fused to the 3’ half of
pyrG.
Overlap PCR using AA20, CC61, CC62 and AA53.
Amplify using AA20 and AA53. |
|
pAA5962 |
MT1612 |
barA |
|
pTW7703 |
ptrA
SK+
|
A. oryzae ptrA PCR amplified from PTRII using primers NN61 and NN62 and cloned into
the SmaI
site of pBluescript II SK+. |
|
pTW7704 |
ptrA
SK+
|
Same as pTW7703
with insert in opposite orientation. |
|
pTW7705 |
ptrA
SK+ backbone |
A. oryzae ptrA PCR amplified from PTRII using primers NN61 and NN62 and blunt cloned
into the
SspI site of the pBluescript II SK+
backbone. |
|
pDG18 |
trpC(p)::hv-tk::trpC(t) |
Contains the
hv-tk
gene encoding herpes simplex virus thymidine
kinase under the control of the
A. nidulans trpC promoter and terminator
(Gardiner and Howlett 2004). |
|
pKB7410 |
pyrG
(2161) pGTE |
pyrG
cloned from strain FRR2161 (pyrG+)
using primers HH81 and II01 and cloned into pGEM-T
Easy. |
|
pKB7411 |
pyrG
(SPM4) pGTE |
pyrG
cloned from strain SPM4 (pyrG-)
using primers HH81 and II01 and cloned into pGEM-T
Easy. |
|
pLS7804 |
pyrG
targeting |
P. marneffei pyrG (missing 60 bp of coding region) amplified with QQ39 and II01,
digested with
EcoICRI
and cloned into the
SspI
sites of the pBluescript II SK+
backbone. |
|
pHB7612 |
P. marneffei niaD |
niaD
was PCR amplified using KK77 and KK78 and cloned
into pBluescript II SK+. |
|
pHB7613 |
niaD
targeting |
EcoRI
fragment deletion to remove the 5’ 546 bp of
niaD to allow targeted integration. |
|
pHB7615 |
niaD
targeting SK+ backbone |
2.8 kb
EcoRI/HindIII fragment from pHB7613 was made blunt ended with
Klenow and cloned into the
SspI
sites in the pBluescript II SK+ backbone. |
|
pHB6104 |
areA |
Full length
areA
gene (Bugeja
et al, 2012b) |
|
pHB7186 |
areA
targeting |
2.6 kb
EcoICRI/EcoRV fragment from pHB6104 cloned into the
SspI
sites of the pBluescript II SK+
backbone. |
|
pSJ7351 |
wA
pGTE |
8.5 kb coding
region of
wA amplified using HH45 and HH46 and cloned
into pGEM-T Easy. |
|
pSB7364 |
wA
targeting (pyrG+) |
A
NotI
fragment of a 1.7 kb
wA PCR
(amplified from pSB7351 using II08 and II09) was
cloned into pALX223. |
|
pDONRTM221 |
Gateway cloning
vector |
Invitrogen
|
|
pHW7711 |
pDONRTM221::pyrG |
Bugeja et al, 2012 |
|
pHW7856, pHW7857. |
pDONRTM221
::pyroA
|
Inverse PCR of
pDONRTM221 with the MM8 and MM9
primers ligated to the
pyroA
fragment from pAA7331.
pyroA
selectable marker available in both
orientations. |
|
pHW7771, pHW7772 |
pDONRTM221
::barA
|
Inverse PCR of
pDONRTM221 with the MM8 and MM9
primers ligated to the 1443 bp
EcoRV barA
fragment from pAA5962.
barA
selectable marker available in both
orientations. |
|
pHW7773,
pHW7774 |
pDONRTM221
::riboB
|
Inverse PCR of
pDONRTM221 with the MM8 and MM9
primers ligated to a 2663 bp
AnriboB
PCR product from pPL3 amplified using the
primers QQ11 and QQ12.
AnriboB selectable marker available in both orientations. |
|
pMP7742 |
pDONRTM221
::ptrA |
Inverse PCR of
pDONRTM221 with the MM8 and MM9
primers ligated to an
EcoICRI/EcoRV
ptrA fragment from pTW7703. |
|
pMP7601 |
mCherry pGTE |
mCherry
PCR amplified from LO1945 (Supplied by B. Oakey)
using LL09 and LL10 and cloned into pGEM-T Easy. |
|
pMP7602 |
H1
pGTE |
Histone H1 PCR amplified from
FRR2161 genomic DNA with LL11 and LL12, which
adds a 5’
BamH1 site and a 3’
SpeI site, removes
the stop
codon and contains 1.7 kb
of
promoter. |
|
pMP7605 |
H1(p)::H1::mCherry
pyrG |
BamHI/SpeI
pMP7602 ligated to
BamHI/SpeI
pALX223. This clone was digested with
SpeI/XbaI
and ligated to
SpeI/XbaI
fragment from pMP7601. |
Table 5. Plasmids for use in the selectable
marker systems
|
Selectable marker |
Plasmid |
Description |
Reference |
|
pyrG |
pAA4707 |
A. nidulans pyrG |
(Oakley et
al. 1987a) |
|
|
pAB4626 |
A. nidulans pyrG blaster cassette |
(Borneman
et al. 2001) |
|
|
pHW7711 |
GatewayTM
plasmid containing
A.
nidulans pyrG blaster cassette flanked by
attP
sites |
(Bugeja et
al, 2012) |
|
|
pSB7364 |
wA
targeting construct contains
A.
nidulans
pyrG
blaster cassette |
This study |
|
|
pLS7804 |
pyrG
targeting construct |
This study |
|
niaD |
pSTA14 |
A. oryzae niaD |
(Unkles et
al. 1989) |
|
|
pHB7613, pHB7615 |
niaD
targeting construct |
This study |
|
riboB |
pPL3 |
A. nidulans riboB gene |
(Oakley et
al. 1987b) |
|
|
pHW7773,
pHW7774 |
GatewayTM
plasmid containing
A.
nidulans riboB gene flanked by
attP
sites |
This study |
|
pyroA |
p14 |
A. nidulans pyroA gene |
(Osmani et
al. 1999) |
|
barA |
pSM6287 |
A. oryzae barA gene |
(Nayak et
al. 2006) |
|
|
pHW7771, pHW7772 |
GatewayTM
plasmid containing
A. oryzae
barA gene flanked by
attP
sites |
This study |
|
ptrA |
pTW7703-7705 |
A. oryzae ptrA gene |
This study |
|
|
pMP7742 |
GatewayTM
plasmid containing
A. oryzae
ptrA gene flanked by
attP
sites |
This study |
|
barA/hv-tk |
pHW7591 |
Plasmid
containing
A. oryzae barA for positive selection and
hv-tk
for negative selection |
This study |
|
areA |
pHB7186 |
areA
targeting construct |
This study |
Pyrithiamine resistance was first developed as a dominant
selectable marker in A. oryzae and has
since been shown to be effective in a number of filamentous fungi
(Kubodera
et al. 2000; Kubodera et al. 2002).
Pyrithiamine is a thiamine analogue, that binds to thiamine pyrophosphate
riboswitches, small RNA elements that bind thiamine pyrophosphate to regulate
the expression of genes required for the biosynthesis and transport of thiamine,
an essential cofactor (Sudarsan
et al. 2005). Pyrithiamine
resistance can also be utilized as a dominant selectable marker for
transformation in P. marneffei as
ptrA containing plasmids (Table 5)
confer resistance to pyrithiamine, with transformants selected on 0.1-0.2 mg mL-1
of pyrithiamine hydrobromide (Sigma)(Table 2). Occasionally, a low number of
spontaneously resistant colonies can arise during transformation without the
addition of exogenous DNA.
Selectable marker
plasmids facilitating positive/negative selection
In circumstances where constructs may be transiently
required, certain selectable markers may also be used for negative selection of
constructs. This has been demonstrated previously for recycling of the
pyrG selectable marker (Borneman
et al. 2001). Both the
pyrG and
niaD genes facilitate negative selection, in addition to positive
selection, since mutations cause resistance to the toxic compounds
5-fluoroorotic acid (5-FOA) or chlorate, respectively (Table 2). A new construct
has been developed which can also be used for both positive and negative
selection (pHW7709, Table 5). This construct contains the previously described
barA gene, used as a positive
selectable marker and the ‘dominant’ Herpes Simplex virus thymidine kinase
encoding gene (hv-tk) as a negative
selectable marker, which confers sensitivity to the toxic thymidine analogue
5-fluorodeoxyuridine (FDU) (Table 2 and 5) (Sachs
et al, 1997; Ahuja and Punekar 2008;
Gardiner and Howlett 2004; Gill and Eisenberg 2001). To counter-select
against hv-tk, strains are plated on
medium containing 5 µM 5-fluorodeoxyuridine (FDU)(Sigma). Southern blot
hybridisation analysis should be used to confirm loss of the constructs
containing the negative selectable markers, as opposed to point mutations that
could result in the same phenotype, albeit at a lower frequency.
Targeted integration
of plasmids
Targeted integration of constructs at specific loci offers
many advantages over non-specific ectopic integration by overcoming possible
copy number and position effects. A series of targeting constructs were
generated to allow for the integration of plasmids at known genomic locations,
including pyrG,
areA, niaD and wA (Table
6). When a recipient strain is transformed with the appropriate targeting
plasmid, a single homologous recombination event leads to the integration of the
plasmid thus restoring gene function (pyrG,
areA and niaD) or resulting in a
visible phenotype (wA) (Figure 1).
Ectopic integration will not result in these selected phenotypes.
The plasmids used for targeted integration at
pyrG, niaD or
areA all contain a portion of the
selectable marker cloned into the SspI
sites of the pBluescript II SK+ backbone to permit blue/white
screening to be used when additional DNA fragments are cloned into the
polylinker (Table 4).
The pyrG-
and niaD- mutations in
strain SPM4 (G147) were identified by sequencing of the PCR products spanning
these genes (Table 6). Plasmids for pyrG
and niaD targeting lack the start
codon of these genes but contain the regions that span the loss-of-function
point mutations. Thus, a single crossover event between the start codon and the
mutated region of the genomic allele leads to integration of the plasmid and
restoration of gene function, that is the ability to grow in the absence of
uracil or on nitrate as a sole nitrogen source (Table 2).
Table 6. P.
marneffei gene targeting regimes
|
Gene targeted |
Recipient strain |
Nature of
mutation in recipient strain |
Region of gene
sequence included for targeting |
|
pyrG |
pyrG- |
L155V and I156T
in the
decarboxylase domain |
Contains all but
first 60 bp of
pyrG
coding sequence (pLS7804) |
|
wA |
Any with green conidiationa
|
N/A |
Internal portion
(+19 to +932) of the
wA
coding sequence
(pSB7364) |
|
areA |
areA∆DBD |
Deletion of DNA
binding domain |
Contains 3’ half
of areA
including DNA binding domain but lacks the START
codon (pHB7186) |
|
niaD |
niaD- |
N293K and K513N |
Contains all but
first 546 bp of
niaD
coding sequence (pHB7615) |
Additionally, the
areA gene, encoding the GATA transcription factor required for growth on
non-preferred nitrogen sources, was also developed as a locus for targeted
integration (Bugeja et al, 2012b). This locus displays a high-rate of homologous
integration and has been modified such that the DNA-binding domain has been
deleted (areA∆DBD),
resulting in loss of gene function, yet the majority of the coding region is
still intact. A plasmid containing the 3’ half of
areA, including the DNA-binding domain is used for targeted
integration (Table 6). When areA∆DBD
strains are transformed with the areA
targeting plasmid, a single crossover event integrates the plasmid at
areA thus restoring the ability to
utilise non-preferred nitrogen sources (Table 2, Figure 1). Screening
integration events by Southern blot analysis is crucial as a double crossover or
gene conversion events are also possible and will lead to
areA+ without integrating the entire plasmid (Figure 1).
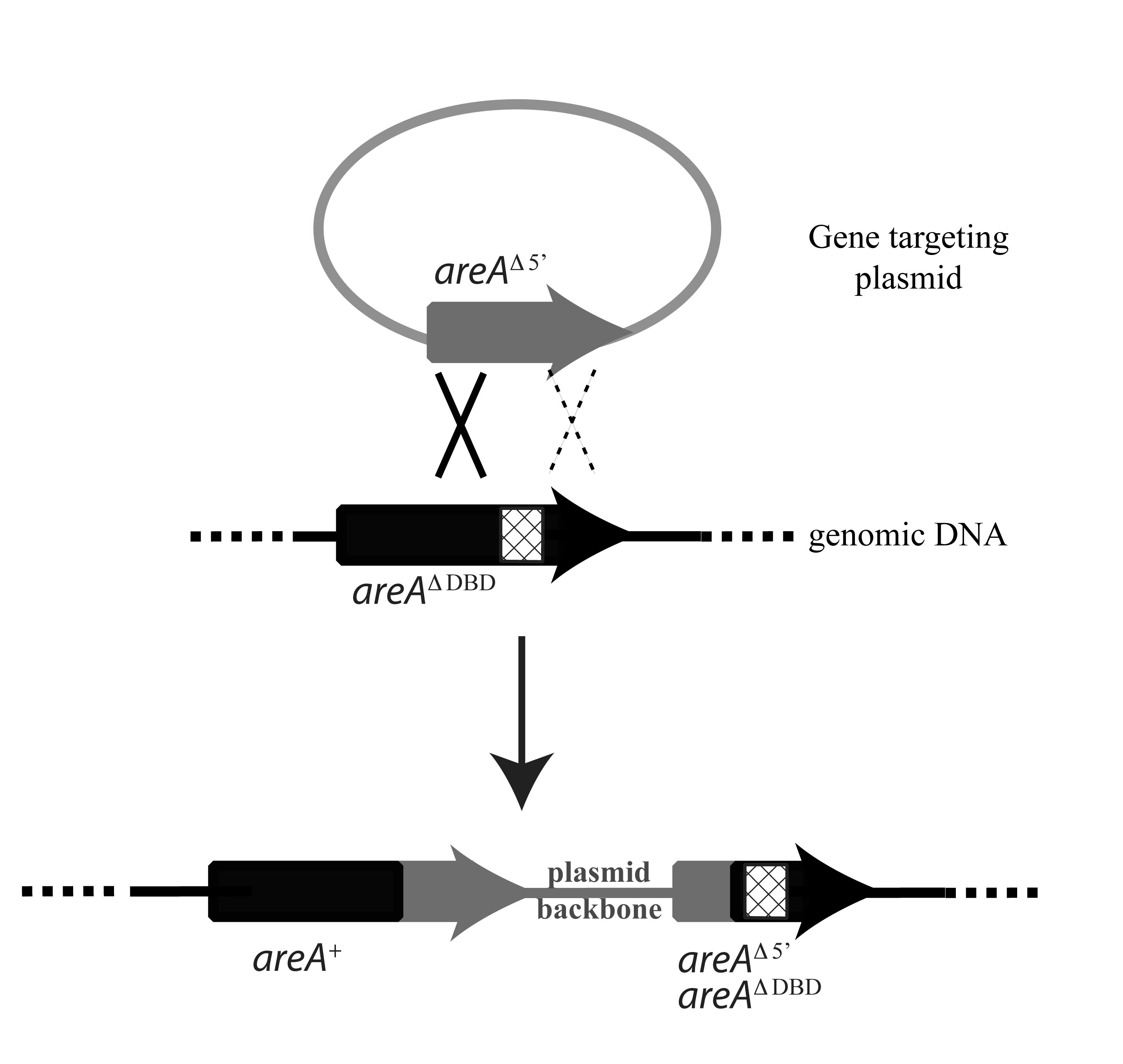
Figure 1. Targeting to the
areA locus in the areA∆DBD strain
The plasmid for gene targeting to
the areA locus contains a 5’ truncated
allele of the areA gene (grey
shading). When areA∆DBD
strains are transformed with the areA
targeting plasmid, a single recombination event will integrate the plasmid into
the genomic region containing the areA∆DBD
locus (black shading) via a single crossover event (solid cross) in the
homologous region 5’ of the DBD deletion (hatched box). This will regenerate a
wild-type areA+ gene thus
restoring the ability to utilise nitrite as the sole nitrogen source, in
addition to, a copy of areA that
contains both the 5’ truncation and the DBD deletion flanking the integrated
vector sequences. The dashed cross depicts an alternate homologous recombination
event that may also occur to regenerate a wild-type
areA+ gene without
integration of the vector sequences.
The polyketide synthase encoding gene,
wA (pksP), is required for DHN melanin
synthesis during asexual development, resulting in pigmentation of the asexual
spores (conidia), which can be detected visually at the colony level
(Mayorga and Timberlake 1990; Mayorga and
Timberlake 1992) (Table 6). The P.
marneffei wA targeting construct contains an internal portion of the
wA coding sequence, in addition to the
A. nidulans pyrG selectable marker
(Table 4). Transformants of pyrG-
recipient strains are selected for uracil prototrophy (pyrG+), and secondarily screened for a white conidial
phenotype indicating that the construct has integrated via a single cross over
event at the wA locus resulting in
gene disruption (Table 6). It should be noted that
A. fumigatus pksP (orthologous to
wA) mutants display decreased virulence in a mouse model of
aspergillosis and P. marneffei
disruption mutants also have attenuated virulence
(Jahn et al. 2000; Jahn
et al. 2002; Langfelder et al.
1998; Woo et al. 2010).
Therefore P. marneffei
wA targeting should not be utilized in
strains that will be subsequently tested for virulence attributes.
Selectable markers
available for the generation of deletion constructs using a GatewayTM
cloning system
A pipeline for the cloning and functional characterization
of genes in P. marneffei utilizing a
GatewayTM cloning system to facilitate the rapid generation of gene
deletion constructs has been developed (Bugeja
et al, 2012). This approach uses a PCR and recombination
based system where the flanking regions of genes to be deleted are amplified by
inverse PCR to incorporate attB
recognition sequences, which facilitates integration of a selectable marker by
in vitro recombination with
corresponding attP sequences. GatewayTM
plasmids containing A. nidulans pyrG
(pHW7711; Figure 2A), riboB (pHW7771
and pHW7772; Figure 2B), pyroA
(pHW7856 and pHW7857, Figure 2C) and A.
oryzae barA (pHW7773 and pHW7774;
Figure 2D) and ptrA (pMP7742; Figure
2E) have been constructed to allow the rapid generation of deletion constructs
(Table 4). These plasmids (which confer kanamycin resistance in
E.coli) have been engineered to contain the selectable marker gene
flanked by attP1 and
attP2 sites. The nature of the
selectable markers means that these constructs can also be used in fungi other
than P. marneffei.
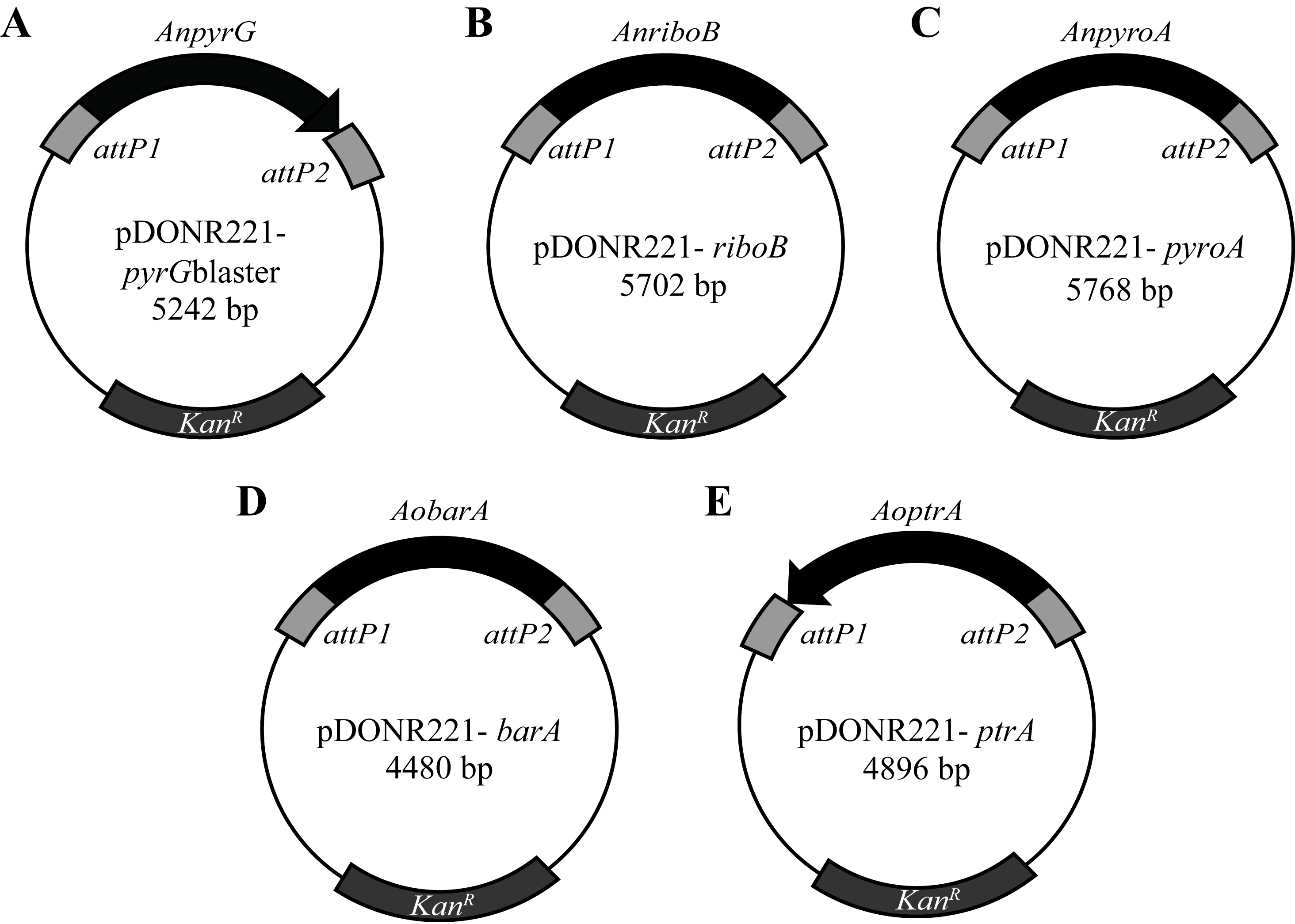
Figure 2. Plasmids
for the generation of deletion constructs using a GatewayTM cloning
system
Plasmids contain kanamycin resistance (KanR) and
a selectable marker flanked by attP1
and attP2 sites. Selectable markers: A.
A. nidulans pyrG
(pHW7711), B. riboB (pHW7771),
C. pyroA (pHW7856) and D.
A. oryzae
barA (pHW7773) and E.
ptrA (pMP7742). Plasmids containing riboA,
pyroA and barA are available with
selectable markers in both orientations.
Tools for
microscopic visualization of P. marneffei
P. marneffei
mutants generated using the molecular tools described above are commonly
characterized for morphological defects microscopically by observing hyphae,
conidiophores and yeast cells.
A number of cellular
stains and fluorescent fusion proteins can be used to allow microscopic
visualization of the cell membrane or wall and nuclei. For microscopic
visualization, P. marneffei
strains can be grown as liquid cultures in shake flasks or microtitre plates, or
on slides covered with a thin layer of solid medium with one end resting in
liquid medium (Borneman
et al. 2000). When required
cells can be fixed by soaking them
in a solution of 4% para-n-formaldehyde in PME (50 mM PIPES pH 6.7, 1 mM MgSO4,
20 mM EGTA) for 30 minutes, followed by two 5 minute PME washes.
P. marneffei
cell membranes can
be visualized using the lipophylic membrane dye FM4-64 (Invitrogen) and is
performed by immersing unfixed slides in 25
mM
FM4-64 (suspended in water) for 15 minutes at room temperature, washing and
mounting in water.
In the wild type,
FM4-64 staining is observed around the cell periphery, surrounding vesicles at
the hyphal apex, as a crescent at the presumptive Spitzenkorper and as
transverse membranes partitioning the hyphae into separate cellular compartments
at septation sites (Figure 3A and B). Membrane sterols can be visualized using the
fluorescent polyene macrolide stain, filipin, which specifically intercalates
into sterol-rich membranes (Van Leeuwen
et al. 2008). Sterol staining
is performed by immersing unfixed cells in 5 mL of 25
mg mL-1 filipin (stock 1 mg mL-1 in
DMSO, Sigma) for 5 minutes, followed by washing with liquid medium and
visualization under UV. In wild
type, ergosterol staining is observed concentrated at the hyphal apex, at the
plasma membrane including at septa and as spots along the length of the hyphae
(Figure 3C and D).
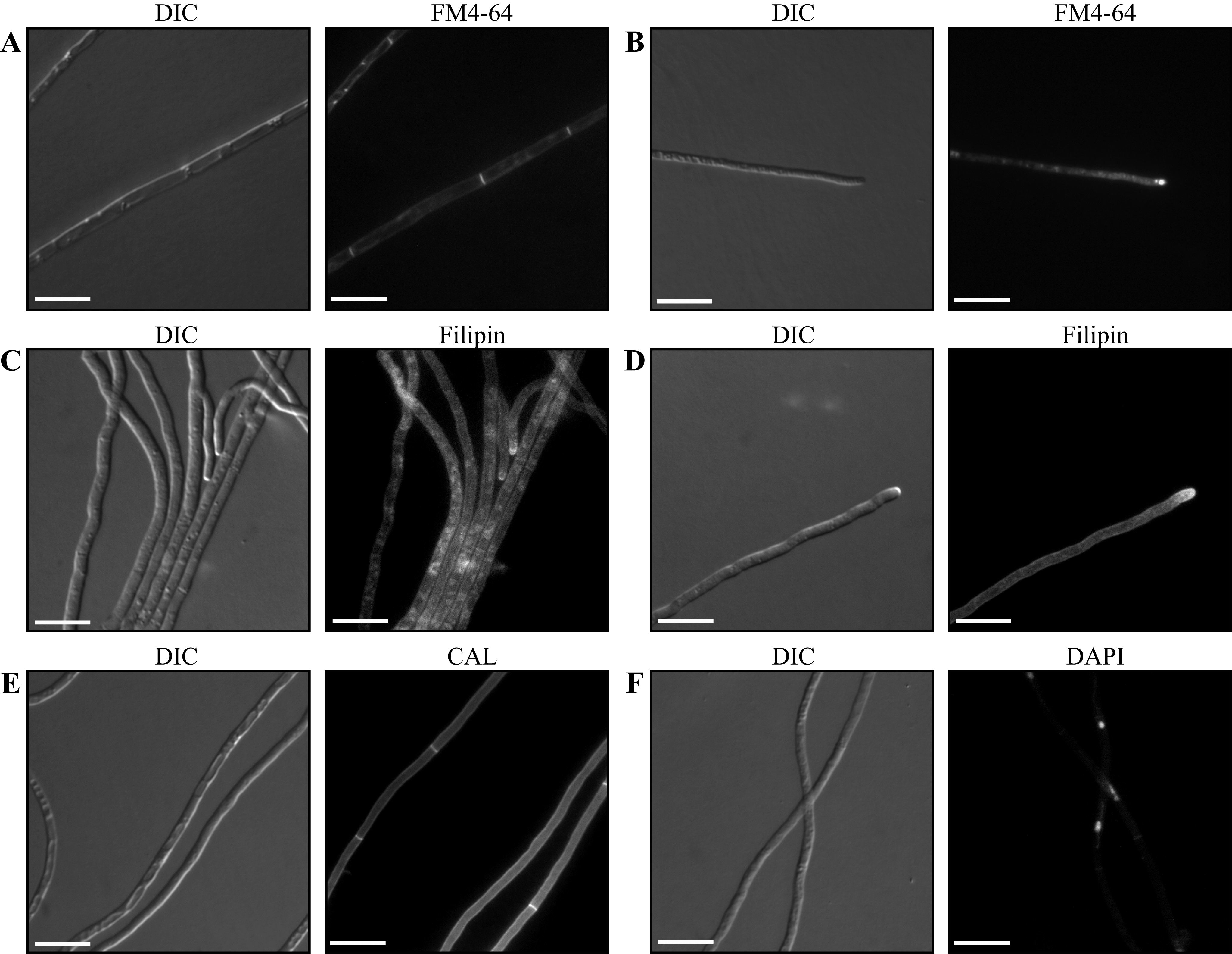
Figure 3.
Microscopic visualization of P. marneffei
Wild type
P. marneffei grown for 3 days at 25°C and stained with FM4-64
(A-B), Filipin (C-D), Calcofluor white (CAL)(E) or DAPI (F). Images were
captured using differential interference contrast (DIC) or with epifluorescence
to observe fluorescent stains. Scale bars, 20
mm.
Fluorescent brightener 28 (calcofluor white, Sigma) and
FITC conjugated lectin (wheat germ agglutinin, WGA, Molecular Probes) can be
used to visualize cell walls under UV
(Figure 3E). Calcofluor is a non-specific fluorochrome which binds both
cellulose and chitin in fungal cell walls, whereas, WGA specifically detects
glycoproteins containing ß(1-4)-N-acetyl-D-glucosamine. For calcofluor white
staining, 1 µL of a 1 µg µL-l calcofluor white solution (suspended in
water) is added directly to 5 µL of Tween 80 on the microscope coverslip prior
to mounting. A modified protocol for WGA staining can be performed on live or
fixed cells (Robin et al. 1986). Prior
to staining, slides are incubated for 5 minutes in PME, 15 minutes in PME with 1
µg µL-1 BSA and then washed in PME. A 5 µL drop of a 5 µg µL-l
WGA solution (suspended in water) is added to the coverslip and slides are
incubated in the dark for 30 minutes, before being washed with PME and mounted.
Under UV light nuclei can be observed in fixed
P.
marneffei cells stained with either 4,6-diamidino-2.phenylindole (DAPI) or
Hoescht 33342 (Figure 3F). 1 µL of a 1 µg
µL-l solution of either stain (suspended in water) is added directly
to 5 µL of Tween 80 on the coverslip prior to mounting. Nuclei can also be
visualized using the HI::mCherry
construct (pMP7605), which contains a fusion between the
Histone HI and the mCherry fluorescent
protein encoding gene (Table 4). A
DligD pyrG-
HI::mCherry strain is also available
as a transformation recipient strain (Table 2).
Combined with strains engineered for homologous
recombination of exogenous DNA, the constructs for ectopic or site-specific
integration and rapid generation of gene deletion constructs described in this
study will greatly facilitate rapid and efficient analysis of gene function in
P. marneffei and are available through
the Fungal Genetics Stock Center (FGSC).
Acknowledgments
This work was supported by grants from the National Health
and Medical Research Council of Australia and the Howard Hughes Medical
Institute.
References
Ahuja, M., Punekar, N.S.,
2008. Phosphinothricin resistance in
Aspergillus niger and its utility as a selectable transformation marker.
Fungal Genet Biol. 45, 1103-10.
Avalos, J.,
et al., 1989. Bialaphos resistance as
a dominant selectable marker in Neurospora
crassa. Curr Genet. 16, 369-72.
Borneman, A.R.,
et al., 2000. The
abaA homologue of Penicillium
marneffei participates in two developmental programmes: conidiation and
dimorphic growth. Mol Microbiol. 38,
1034-47.
Borneman, A.R.,
et al., 2001. An
STE12 homolog from the asexual, dimorphic fungus
Penicillium marneffei complements the
defect in sexual development of an
Aspergillus nidulans steA mutant. Genetics. 157,
1003-14.
Bugeja, H.E.,
et al., 2012.
Tools for high efficiency genetic manipulation of the human pathogen
Penicillium marneffei. Fungal Genetics Biol. In press.
Bugeja, H.E.,
et al., 2012b.
AreA controls nitrogen source utilisation during both growth programs of the
dimorphic fungus Penicillium marneffei.
Fungal Biol. 116, 145-54.
Gardiner, D.M., Howlett,
B.J., 2004. Negative selection using thymidine kinase increases the efficiency
of recovery of transformants with targeted genes in the filamentous fungus
Leptosphaeria maculans. Curr Genet. 45,
249-55.
Gill, H.S., Eisenberg, D.,
2001. The crystal structure of phosphinothricin in the active site of glutamine
synthetase illuminates the mechanism of enzymatic inhibition. Biochem. 40,
1903-12.
Jahn, B.,
et al., 2000. Interaction of human phagocytes with pigmentless
Aspergillus conidia. Infect Immun. 68,
3736-9.
Jahn, B.,
et al., 2002. PKSP-dependent
reduction of phagolysosome fusion and intracellular kill of
Aspergillus fumigatus conidia by human
monocyte-derived macrophages. Cell Microbiol. 4, 793-803.
Kubodera, T.,
et al., 2000. Pyrithiamine resistance
gene (ptrA) of
Aspergillus oryzae: cloning, characterization and application as a
dominant selectable marker for transformation. Biosci Biotechnol Biochem. 64,
1416-21.
Kubodera, T.,
et al., 2002. Transformation of
Aspergillus sp. and
Trichoderma reesei using the pyrithiamine resistance gene (ptrA)
of Aspergillus oryzae. Biosci Biotechnol Biochem. 66,
404-6.
Langfelder, K.,
et al., 1998. Identification of a
polyketide synthase gene (pksP) of
Aspergillus fumigatus involved in
conidial pigment biosynthesis and virulence. Med Microbiol Immunol. 187,
79-89.
Mayorga, M.E., Timberlake,
W.E., 1990. Isolation and molecular characterization of the
Aspergillus nidulans wA gene.
Genetics. 126, 73-9.
Mayorga, M.E., Timberlake,
W.E., 1992. The developmentally regulated
Aspergillus nidulans wA gene encodes a polypeptide homologous to polyketide
and fatty acid synthases. Mol Gen Genet. 235,
205-12.
Nayak, T.,
et al., 2006. A versatile and
efficient gene-targeting system for
Aspergillus nidulans. Genetics. 172,
1557-66.
Oakley, B.R.,
et al., 1987a. Cloning, mapping and
molecular analysis of the pyrG
(orotidine-5'-phosphate decarboxylase) gene of
Aspergillus nidulans. Gene. 61,
385-99.
Oakley, C.E.,
et al., 1987b. Cloning of the
riboB locus of
Aspergillus nidulans. Gene. 53,
293-8.
Osmani, A.H.,
et al., 1999. The extremely conserved
pyroA gene of
Aspergillus nidulans is required for pyridoxine synthesis and is
required indirectly for resistance to photosensitizers. J Biol Chem. 274,
23565-9.
Robin, J.B.,
et al., 1986. Rapid visualization of
three common fungi using fluorescein-conjugated lectins. Invest Ophthalmol Vis
Sci. 27, 500-6.
Sachs, M.S.,
et al, 1997. Expression of
herpes virus thymidine kinase in
Neurospora crassa. Nucleic Acids Res. 25, 2389-95.
Sudarsan, N.,
et al., 2005. Thiamine pyrophosphate
riboswitches are targets for the antimicrobial compound pyrithiamine. Chem Biol.
12, 1325-35.
Unkles, S.E.,
et al., 1989. Transformation of
Aspergillus niger with the homologous
nitrate reductase gene. Gene. 78,
157-66.
Van Leeuwen, M.R.,
et al., 2008. Filipin is a reliable
in situ marker of ergosterol in the
plasma membrane of germinating conidia (spores) of
Penicillium discolor and stains intensively at the site of germ tube
formation. J Microbiol Methods. 74,
64-73.
Woo, P.C.,
et al., 2010. High diversity of
polyketide synthase genes and the melanin biosynthesis gene cluster in
Penicillium marneffei. FEBS J. 277,
3750-8.