Optimized primers and other critical
conditions for efficient fusion PCR to generate knockout vectors in filamentous
fungi.
Nicholas Harrison1, Brad Cavinder
1,2, Jeffrey P.
Townsend3, and
Frances Trail1, 4
1 Department of Plant Biology, Michigan State
University, East Lansing, MI 48824. 2Current address: Department of
Plant Pathology, University of California, Riverside. Email: brad.cavinder@ucr.edu. 3Department
of Ecology and Evolutionary Biology, Yale University, New Haven CT. Email: Jeffrey.Townsend@Yale.edu. 4Corresponding
author: trail@msu.edu
Fungal Genetics Reports 60:1-
(pdf)
Methods to streamline functional studies
of large numbers of genes are essential to fully utilize the significant
genomic resources now available for fungi.
Fusion PCR is often used to join pieces of DNA together, particularly in
the construction of DNA fragments for gene replacement in fungi. Here we
present high-efficiency primers which reliably direct fusion and amplification
to generate constructs for gene knockouts.
Introduction
FPCR-based
gene replacement strategies have been shown to be useful in multiple
fungi, including Saccharomyces
cerevisiae, Cochliobolus
heterostrophus, Fusarium graminearum, and Aspergillus nidulans (Catlett et
al. 2003, Szewczyk et al. 2006;
Cavinder et al. 2011, Cavinder and
Trail 2012). Previously, it was reported
that the efficiency of targeted gene replacement could be enhanced by employing
a split-marker approach where regions flanking the gene of interest are fused
to overlapping partial segments of a selectable marker (Figure 1; Fairhead et al. 1998, Catlett et al. 2003). For a split marker gene replacement two
constructs are generated: a 5’ constuct, containing an upstream flanking region
and a 5’ segment of the selectable marker, and a 3’ construct, containing a
downstream flanking region and a 3’ segment of the selectable marker (Figure 1A
and B). These two constucts are used for
transformation of the organism in which three crossovers occur: both flanking
regions in the genome crossover with their complementary sequences in the two
contructs, and the overlapping regions of partial marker segments crossover to
form the complete selectable marker (Figure 1C). Thus, the gene of interest is completely
replaced with the selectable marker.
FPCR was first demonstrated in filamentous fungi as a tool for gene
replacement by Catlett et al. (2003).
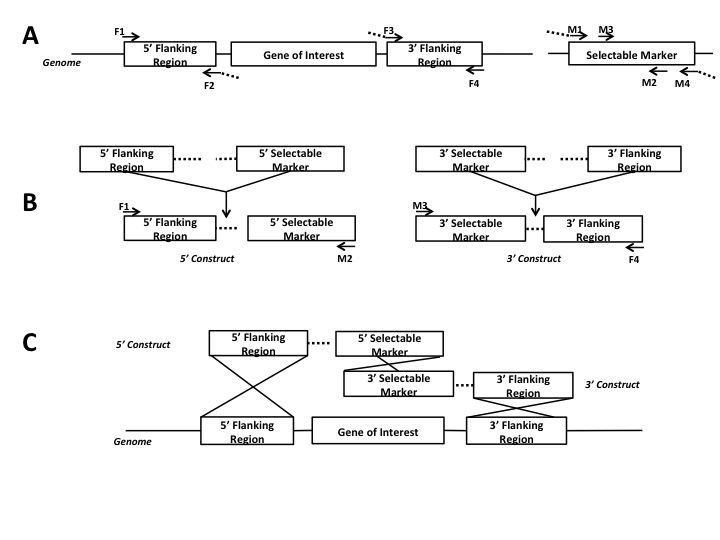
Figure 1. Diagram of a
split-marker gene replacement strategy using fusion-PCR
(A) FPCR step 1 -
amplification of flanking regions and partial marker segments. Dotted tails indicate the overlap
sequences on selected primers which allow fusion in step 2. (B) FPCR step 2 - flanking regions are
fused to the marker segments at the overlap sights, forming two constructs.
The fusion products are then amplified.
(C) Transformation. Three
crossovers occur during the gene replacement. Each genomic flanking region
crosses over with its complementary sequence in the FPCR constructs. The complementary portions of the marker
segments crossover to make the completed selectable marker.
Because
FPCR requires only PCR primers and reagents, cloning procedures based on this
method can often be quicker, easier, or less expensive than other cloning strategies such as
restiction digests and ligations or commercial gene synthesis (Ellis, Adie, and
Baldwin 2011). Nonetheless, the
efficiency of FPCR reactions can be sensitive to conditions such as primer
concentration, template concentration, annealing temperature, the size of the
segments to be fused, and the nature of the chimeric overlap sequences (Bryksin
and Matsumura 2010, Chai-aim et al. 2009). The latter, in particular, can have a serious
impact on the success of FPCR because using native sequences as the chimeric
overlapping sequences can frequently
result in poor fusion or even none at all (Chai-aim et al. 2009, Chai-aim et al. 2012).
Little has been published on the effects different overlapping sequences have
on fusion, although previous reports indicated that 15 base pair overlap sequences
rich in repeating G and C nucleotides resulted in excellent fusion with broad
applicability (Chai-aim et al. 2009,
Chai-aim et al. 2012). Others, however, have suggested that
sequences containing high G/C ratios and palindromic elements cause problems in
PCR and FPCR reactions (Ellis, Adie, and Baldwin 2011, Zhao et al. 2011). We examined the design of high efficiency
primers in the generation of split-marker gene replacement constructs, using Fusarium graminearum as our target organism. Our results identify primer design strategies
and PCR conditions that optimize efficiency in generating gene replacement
constructs via FPCR.
Methods
Strains and Primer Design
All studies were
performed on Fusarium graminearum strain
PH-1 (Trail and Common 2000),
which was the strain on which the genomic sequence was based (Cuomo et al.,
2007). Genomic sequences for PH-1 were
obtained from the MIPS F. graminearum genome
database (http://mips.helmholtz-muenchen.de/genre/proj/FGDB/). Four genes were
chosen for comparison of the different strategies of gene disruption. They are designated in the MIPS Fusarium graminearum genome database as
FGSG_4001, FGSG_4180, FGSG_7376, and FGSG_16930. Eight primers were used to generate FPCR constructs for each gene
target (Figure 1). Primers F1 and F2
amplified the upstream flanking region while primers F3 and F4 amplified the
downstream flanking region. The flanking
amplicons were 500-700 base pairs in length, amplified from genomic DNA (FGSC9075,
NRRL31084). All primer sequences were screened for hairpin
formation and 3’ end primer dimmer formation by the IDT OligoAnalyzer online
program (http://www.idtdna.com/analyzer/Applications/OligoAnalyzer/). Primer sequences were selected with G/C
contents between 40-60%, melting temperatures of 60˚C, and 1-2 base pair
G-C clamps at terminal ends.
The
E. coli hygromycin phosphotransferase
gene hph conferring resistance to
hygromycin was used as the selectable marker, under the control of the trpC
promoter and terminator from A. nidulans (amplified
from plasmid pCB1004; Carroll, Sweigard and Valent 1994) and was amplified in
two overlapping pieces using primers M1x M2 and M3 x M4 (Figure 1A). M1 and M2 primed the 5’ selectable marker
segment, while M3 and M4 primed the 3’ selectable marker segment. There was a 1.1 kB overlap between the 5’ and
3’ selectable marker segments for mediating homologous recombination during
transformation (Figure 1C).
Fusion
PCR
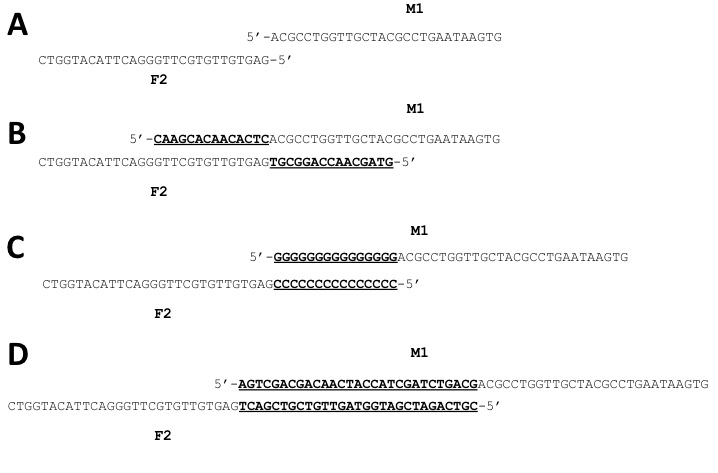
Figure 2. Examples of F2 and M1 primers for
gene target FGSG_4001 (A) Primer stems only (no
overlaps). (B) The bold/underlined sequences were added
to the primer stems to create NSOs that incorporate the sequence of each
primer into the overlap. (C) 15G/15C overlap added to primer
stems. (D) 5HS overlap added to
primer stems.
Generation of knockout
constructs by FPCR was accomplished in a two-step procedure. In Step 1, marker and flanking regions were
amplified from pCB1004 and F. graminearum
genomic DNA templates, respectively, using Phusion DNA polymerase (New England
Biolabs, which has been previously shown to optimize the efficiency and error
rate of FPCR reactions (Bryksin
and Matsumura 2010). The 5’ flanking
regions were amplified with F1 and F2 primers, the 3’ flanking region with F3
and F4 primers, the 5’ marker segement with M1 and M2 primers, and the 3’
marker segment with M3 and M4 primers (Figure 1A). These reactions were mixed as per
manufacturer’s directions. An initial denaturation at 98˚
for 30s - 3 min (longer for genomic DNA, shorter for pCB1004) was followed by
31 cycles of 98 ̊for 10s
(denaturation), 60-64˚ for 30s (annealing), and 72̊C for 50s
(extension). After the final cycle a 10 min final extension at 72˚
finished the program. Although our primers
had melting temperatures of 60̊ C, we tested annealing temperatures of
both 60˚C and 64˚C to accomodate the manufacturer reccomendations for
reactions with the Phusion DNA polymerase.
Step 1 reactions were separated on 0.6% agarose gels. Bands were excised
and purified with the Wizard SV gel and PCR clean-up kit (Promega). The purified amplicons were then used in step
2 for fusion.
In
Step 2, the fusions of 5’ flanking regions to 5’ selectable marker segments,
and 3’ flanking regions to 3’ marker segments were performed (Figure 1B) to produce
the 5’ and 3’ constructs for each gene, respectively. Reactions were first mixed as follows for
each construct: 10 ng-1.2
µg each of flanking region and marker segment DNA (these serve as the
template in this reaction), in a 50 µL with the other components as per
manufacturer’s directions for Phusion DNA polymerase. Because some FPCR-based applications have
been reported to be sensitive to both the concentration of template used in
this step and the concentration ratios of the segments to be fused, we tested
several different ratios and concentrations between 10ng and1.2 µg for
each piece. These reactions were
submitted to 30s at 98˚C initial denaturation followed by 8
cycles of: 10s at 98˚C denaturation, 30s at 64-68˚C annealing, and 1min at 72˚C extension. Once again, the program concluded with a 10 min,
72˚C, final extension time.
Note that the annealing step in this reaction was the annealing of the
overlap sequences between the flanking and marker segments and not the
annealing of primers since none were included in the reaction mix. We also tested reaction conditions where the
overlaps were allowed to both anneal and extend at 72˚C, by simply eliminating the separate annealing step.
Products of the above fusions were used to make the final
merged constructs. The following mix was made: 10 µL of
5X Phusion HF buffer, 0.25 µM final concentration of each primer,
200 µM final concentration of dNTP mix,
0.5 µL of Phusion enzyme, and DNase-free
water up to a total volume of 50 µL.
These mixes were added to each reaction.
The primers used for 5’ constructs were F1 and M2; for 3’ constructs
they were M3 and F4. A final program, to
amplify the fused constructs, was run with a 30s at 98˚C initial denaturation followed by 31 cycles of 98˚C for 10s, 60-64˚C for
30s, and 72˚ C for 1 min. A final
extension of 10 min concluded the program.
These reactions were separated by electrophoresis on 0.6 % agarose gels
and DNA purified with the Wizard SV gel
and PCR clean-up kit.
Transformation
and screening of transformants
Transformation
of F.graminearum strain PH-1 with the
merged constructs was accomplished by the polyethylene glycol/protoplast
method, as previously described (Hallen-Adams, Cavinder, and Trail 2011). Putative transformants were screened on V8
agar amended with 450 µg/mL
hygromycin. Hygromycin-resistant
transformants were then grown in 5-8 mL carboxymethylcellulose (CMC) broth to
produce condia (Cappellini and Perterson 1965).
Spores were germinated on water agar and single-spore isolates were recovered
to ensure each transformant was a true-breeding strain. Mycelia from each strain were grown in 10 mL
YES broth, frozen, and lyophilized.
Genomic DNA was extracted from the lyophilized tissue. PCR amplification of the locus of interest
was then used to determine whether constructs had correctly integrated into the
locus and replaced the target gene.
Results
Evaluation
of conditions affecting the success of amplification by primers
We
compared three strategies for designing primers that would drive merges. In the first strategy (Figure 2B) we used the native
gene sequences (Native Overlap Sequences; NSO) as our overlapping
sequences. Prior to this study, this had
been our method for generating knockouts. The second
strategy (Figure 2C) employed two overlapping sequences containing repeating
strings of G and C nucleotides previously reported to enhance merge efficacy (Chai-aim et al. 2009, Chai-aim et al. 2012).
In
the third strategy (Figure 2D), we developed two novel sequences with a
heterogeneous and non-repeating mix of all four nucleotides. We designed primers using NSO for 11 target
genes, as part of an ongoing gene knockout project, immediately prior to this
study. We chose four additional genes
for targeted replacement to compare the G-C rich and heterogeneous sequence (HS)
overlap methods (Figure 2C and D). The four
genes were FGSG_4001, FGSG_4180, FGSG_7376, and FGSG_16930. Step 1 of FPCR was the amplification (by standard PCR
conditions) of marker and flanking segments (Figure 1A). For all primer pairs, annealing
temperatures of 64°C produced successful amplifications
for the majority of trials as compared to 60°C
(data not shown).
We tested whether
different overlaps had an effect on the success of amplification of the two
flanking regions and the two selectable marker segments for our four chosen
genes. Each amplification reaction
contained one primer without an overlap and one primer with an overlap (Figure
1). Table 1 summarizes the success of these amplification reactions related to
the type of overlap primer used. In
total, 12 out of 20 amplifications using a primer with a G-C rich overlap had
weak amplification or failed to amplify completely. When the G-C rich overlaps on these primers
were replaced by 5HS and 3HS overlaps, all of the reactions returned strong
amplification. Figure 3 shows results
from separation of fragments by agarose gel electrophoresis for these
amplifications. After receiving these
results, we created constucts for 15 additional genes using the 5HS and 3HS
overlaps, with similarly successful results.
In addition, the application of NSOs to amplify flanking segments for 15
target genes, which are part of an ongoing gene knockout project, also resulted
in successful amplification for all segments.
|
Table 1. Summary of results of FPCR step 1 reactions in comparison of
overlaps |
|||
|
Type of overlap |
Results of FPCR step 1
reactions (out of 5 reactions/overlap) |
||
|
Strong Amplification |
Weak Amplification |
No Amplification |
|
|
15C |
3 |
1 |
1 |
|
15G |
0 |
0 |
5 |
|
5CGC |
3 |
1 |
1 |
|
5GCG |
2 |
0 |
3 |
|
5HS |
5 |
0 |
0 |
|
3HS |
5 |
0 |
0 |
Evaluation
of conditions affecting fusion of amplified segments
For
our four genes, the amplified segments of the 5’ flanking regions to 5’
selectable marker segments, and 3’ flanking regions to 3’ marker segments were merged
to produce the 5’ and 3’ constructs for each gene, respectively (Figure 1B). The
resulting fusion products were designated as 5’ and 3’ constructs,
respectively. Since many of the segment amplifications involved primers with
G-C rich overlaps that failed, only 3 pairs of flanking regions and
corresponding marker segments were available to be fused. Of these, all three returned strong bands in
agarose gel electrophoresis with sizes corresponding to completed constructs
(Figure 4). Similarly, all fusion reactions
with 5HS or 3HS overlaps yielded strong bands.
Results of fusions with NSO containing segments were highly variable.
Allowing
overlaps to anneal to each other at 68° in the first PCR program of fusion (using
an 8 cycle program, see Methods section) consistently yielded stronger fusion
than annealing at 64°, resulting in stronger bands corresponding to the desired
product (Figure 4). Removing the
annealing step in this program, and allowing the overlaps to both anneal and
extend at 72°, provided indistinguishable results from programs with the 68°
annealing step. Using more than 8
cycles did not improve the specificity of the fusion, and using more than 10-15
cycles increased the amount of side-reactions as viewed by extra and unexpected
bands on the gels. In the second amplification step (the 31 cycle amplification,
see Methods section) primer annealing
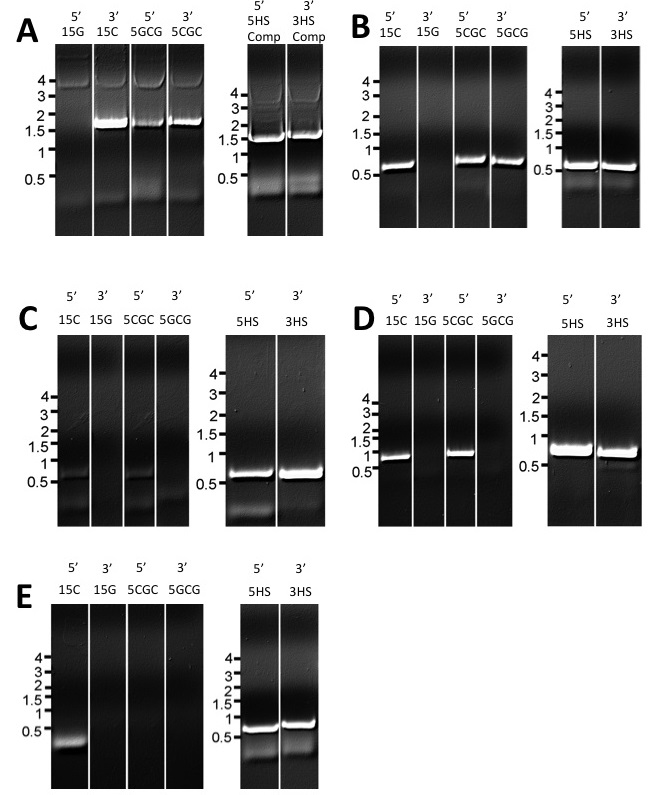
Figure 3. FPCR step 1 amplifications of flanking and marker segments
with different overlaps. 5’ or 3’ indicates the identity of the segment
amplified (e.g. 5’ marker or 3’ marker)
15C, 15G, 5CGC, 5GCG, 5HS, and 3HS indicate the overlap sequence on
the overlap primer used for the amplification. 5HS comp and 3HS comp refer to the 5HS
and 3HS complement sequences on the M1 and M4 primers (A) Marker
segments (B) FGSG_4001 flanking
segments (C) FGSG_4180 flanking
segments (D) FGSG_7376 flanking
segments (E) FGSG_16930 flanking
segments
temperatures of 64°
proved preferable to 60° as viewed by stronger bands when reactions products
were resolved by agarose gel electrophoresis.
We also tested several different concentrations and
ratios of marker segment and flanking region DNA in the first reaction mix of
step 2 for the four genes examined.
Despite concentrations of each piece ranging from 10 ng-1.2 µg and
marker:flanking segment ratios ranging from 12:1 to 1:12, we did not notice a
difference in the success of these reactions between different conditions as
seen by band size during separation on agarose gels.
For the 11 gene targets for which we used the NSO,
immediately prior to this study, only four yeilded at least 2 confirmed gene
replacement strains. However, for
the 15 gene targets (using SH to generate the knockout constructs) we performed
transformation experiments using completed 5’ and 3’ constructs (Figure
1C). Transformations yielded between
5-20 recombinant transformants for each of the 15 genes. We analyzed putative transformants by PCR
amplification to separate ectopic insertional mutants from true deletion
mutants. We screened transformants until
we confirmed at least 2 true deletion strains for 12 of the 15 genes. Three genes yielded only ectopic
transformants. We repeated transformation
experiments three times each for these genes, with similar results, before
concluding that the deletion of these genes may produce lethal mutations. The phenotypic analysis of the knockout
transformants will be published separately.
Discussion
We
observed very inconsistent results when using the native gene sequences in
designing overlaps for deletion constructs.
While some fusions worked quite well, others produced unacceptably low yields
of fusion product and many failed to yield any product. Of 11 genes we attempted to knockout by the
NSO method immediately preceding this study, only four were efficiently knocked
out. This supported the hypothesis that
the specific overlap sequence used can have a significant impact on the success
of FPCR applications. It also indicated that strategies which utilize the same
overlaps each time, regardless of what segments are being fused, may provide an
advantage through their consistency.
We tested two such
strategies, the G-C rich overlaps and the HS overlaps, and indeed found that
they produced consistent fusion across multiple constructs. Nonetheless, with the G-C rich overlaps we
observed other problems which may make them unsuitable for creating gene
replacement constructs. While it had
been previously shown that overlaps such as the 15C/15G and 5CGC/5GCG sequences
promote strong and specific fusion products, we hypotheisized that adding long
repeats of G and C nucleotides to the tail of an oligonucleotide in this way
could cause the formation of primer secondary structures that could inhibit
standard PCR amplification. The great
difficulty we had with amplifications using such primers supports this
hypothesis. Primer synthesis companies
often warn against ordering primers with stretches of 6 or more G nucleotides
in particular, because such oligonucleotides can be extremely difficult to
synthesize reliably, and it is noteworthy that none of the amplifications with
15G primers worked at all. Many of the amplifications
with 15C, 5CGC and 5GCG primers also failed to produce product. The
issues we had in amplifying with these primers continued even when we altered
the annealing temperatures and primer concentrations used. Perhaps more telling, though, is that
amplification with these primers worked every time when the same gene regions
were amplified with overlaps containing a more heterogenous mix of nucleotides
(5HS and 3HS). The G-C overlaps did
facilitate strong and specific fusion as previously reported, although the
heterogeneous sequence overlaps produced fusions of equally good quality. Thus, due to their non-interefering nature in
amplification and promotion of strong fusion, we see our novel 5HS and 3HS
sequences as far more desirable options for FPCR, at least in a low complexity
application like the creation of split-marker gene replacement constructs. Although we designed constructs for F.graminearum, these overlaps and our
overall strategy should work equally well in FPCRs for other fungi that can be
transformed with targeted gene replacement constructs. It is worth noting, however, that if these
overlaps were used for FPCR applications such as gene tagging, which requires
maintenance of the reading frame, they would need to be altered.
We
expected primer and template concentrations to be important factors for the
success of both steps of the FPCR. Surprisingly,
neither seemed to have a significant impact on outcomes. Even varying the ratio of marker segment DNA
to flanking segment DNA between 12:1 and 1:12 seemed to have little effect on
producing strong fusion products. It may
be that since our application represents a fairly simple FPCR, having only one
fusion per reaction and using relatively small and similarly sized segments of
DNA, it is insensitive to such conditions.
This is encouraging when trying to assess the overall utility of this
method because fewer limitations on certain conditions should make these
methods more easily adaptable.
Annealing
temperature had a much more significant effect on amplification. Lower annealing temperatures produced
inferior fusion products with lower yields and less specificity. We found excellent results with amplification
annealing temperatures of 64° and overlap annealing temperatures of 68-72°,
however the optimal temperatures may be lower if an enzyme other than Phusion
is used. The Phusion enzyme is only one
of several high-fidelity polymerases, and another such enzyme may yield
equivalent success. It is our recommendation
that for any FPCR annealing temperatures be raised to the highest level for
which fusion and priming are still possible (although not exceeding the temperature of extension). Higher temperatures increase not only the
specificity of priming, but also the specificity of overlaps binding to one
another in step 2. This is important
because under less specific conditions (with lower temperatures) the DNA
segments being fused may bind to each other in undesirable ways that create
unwanted side products. This effect can be directly seen when the fusion
reactions are run out on a gel, as a large amount of unwanted bands and a lack
of amplification on the desired band.
Using
5HS and 3HS overlaps and higher annealing temperatures, we were able to
consistently produce strong fusion products. While our fusions with the heterogenous
sequence overlaps (and the G-C rich overlaps as well) did have a few weak undesirable
bands (Figure 4), the conditions used apparently did not produce enough interference from side reactions
to inhibit the strong amplification of the desired products. 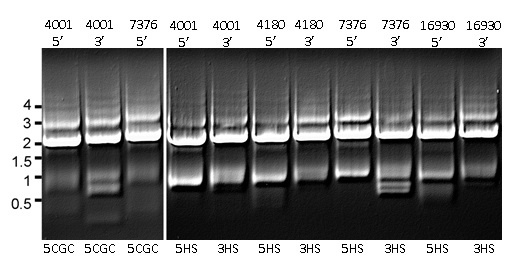
Figure 4. Separation by gel
electrophoresis of merge reactions of amplified segments of the 5’ or 3’ flanking
regions to selectable marker gene using either G-C rich (left panel) or
heterogeneous sequence overlaps (HS, right panel). Numbers at left indicate
size in kB.
Gene designation is indicated above each lane. For each reaction, merge product size is
approximately 2 kB. Products of amplification at 64 C for
both Step 1 and Step 2, and fusion temperature of 68 C in Step 2.
The desired products were easily extracted
from gels with the Promega Wizard SV gel and PCR clean-up kit, with any
possible loss in the quantity/quality of DNA unnoticable in the following
steps. When transformed into F.graminearum
we recovered an excess of recombinant strains for each gene target, which we
confirmed as gene replacements by PCR. Of 24 genes subjected to knockout by the
HS method since this study, 20 have yielded verified knockouts. Because of the
overall speed, low cost, and repeatablity of this method, it should be
exceptionally useful for producing deletion mutants in many filamentous
fungi. Adapting the method we used for F. graminearum to other fungi may be as
easy altering the length of the flanking regions (to match the specific
recombinatorial requirements of each fungus) and using a different selectable
marker if required.
References
Bryksin, A. V.. and,
I. Matsumura. (2010). Overlap extension PCR cloning:a simple and reliable way
to create recombinant plasmids. BioTechniques
48: 463-465.
Cappellini, R.A., and J.L. Peterson. (1965).
Macroconidium formation in submerged cultures by a non-sporulating strain of Gibberella zeae. Mycologia 58:
962-966.
Carroll, A. M., J. A.
Sweigard, and B. Valent. (1994). Improved Vectors for Selecting Resistance to
Hygromycin. Fung. Genet. Newsl. 41:
22.
Catlett, N. L., B.-N.
Lee, O. C. Yoder, B. G. Turgeon. (2003). Split-Marker Recombination for
Efficient Targeted Deletion of Fungal Genes. Fungal Genet. Newsl. 50. 9-11.
Cavinder, B., A. Hamam, R.R. Lewis, and F. Trail.
(2011). Mid1, a mechanosensitive calcium ion channel, affects growth,
development, and ascospore discharge in the filamentout fungus Gibberella zeae. Euk. Cell 10: 832-842.
Cavinder, B., and F. Trail
(2012). The role of Fig1, a component of the Low Affinity Calcium Uptake
System (LACS), in growth and sexual development of filamentous fungi. Eukaryotic Cell 11: 978-988.
Cha-aim, K., H. Hoshida,
T. Fukunaga, and R. Akada. (2012). Fusion PCR via Novel Overlap Sequences. In
Gene Synthesis: Methods and Protocols (pp. 97-110). New York: Springer
Science+Buisiness Media.
Chai-aim, K., T. Fukunaga,
H. Hoshida, and R. Akada. (2009). Reliable fusion PCR mediated by GC-rich
overlap sequences. Gene 434:
43-49.
Cuomo, C.A., U. Gueldener, J.R. Xu, F. Trail, B.G. Turgeon, A. Di Pietro, J.D.
Walton, L.J. Ma, S.E. Baker, M. Rep, G. Adam, J. Antoniw, T. Baldwin, S.
Calvo, Y.L. Chang, D. DeCaprio, L.R. Gale, S. Gnerre, R.S. Goswami, K.
Hammond-Kosack, L.J. Harris, K. Hilburn, J.C. Kennell, S. Kroken, J.K.
Magnuson, G. Mannhaupt, E. Mauceli, H.W. Mewes, R. Mitterbauer, G.
Muehlbauer, M. Munsterkotter, D. Nelson, K. O'Donnell, T. Ouellet, W.H. Qi,
H. Quesneville, M.I.G. Roncero, K.Y. Seong, I.V. Tetko, M. Urban, C.
Waalwijk, T.J. Ward, J.Q. Yao, B.W. Birren, H.C. Kistler. The Fusarium graminearum genome reveals a
link between localized polymorphism and pathogen specialization. (2007). Science
317: 1400-1402.
Ellis, T., T. Adie, and
G. S. Baldwin. (2011). DNA assembly for synthetic biology: from parts to
pathways and beyond. Integr. Biol. 3:
109-118.
Fairhead, C., A. Thierry,
F. Denis, M. Eck, and B. Dujon. (1998). 'Mass-murder' of ORFs from three
regions of chromosome XI from Saccharomyces cerevisiae. Gene 223: 33-46.
Hallen-Adams, H. E., B.
Cavinder, and F. Trail. (2011).
Fusarium graminearum from Expression Analysis to Functional Assays. In Fungal
Genomics: Methods and Protocols (pp. 79-101). New York: Springer
Science+Business Media.
Heckman, K., and L.
Pease. (2007). Gene splicing and mutagenesis by PCR-driven overlap extension.
Nature Protocols 2: 924-932.
Ho, S. N., H. D. Hunt,
R. M. Horton, J. K. Pullen, and L. Pease. (1989). Site-directed mutagenesis
by overlap extension using the polymerase chain reaction. Gene 77: 51-59.
Horton, R. M., H. D. Hunt,
S. N. Ho, J. K. Pullen, and R. L. Pease. (1989). Engineering hybrid genes
without the use of restriction enzymes: gene splicing by overlap extension. Gene 77: 61-68.
Kralicek, A. V., M. Radjainia,
N. A. Mohamad Ali, C. Carraher, R. D. Newcomb, and A. K. Mitra. (2011). A
PCR-directed cell-free approach to optimize protein expression using diverse.
Protein Expression and Purification
80: 117-124.
Maier, F. J., S. Malz,
A. P. Losch, T. Lacour, and W. Schafer. (2005). Development of a highly
efficient gene targeting system for Fusarium graminearum using the disruption
of a polyketide synthase gene as a visible marker. FEMS Yeast Research 5: 653-662.
Trail, F. and R. Common. (2000). Perithecial development by Gibberella zeae: A light microscopy
study. Mycologia 92:130-138
Szewczyk, E., T. Nayak,
C. E. Oakley, H. Edgerton, Y. Xiong, N. Taheri-Talesh, B. R. Oakley. (2006).
Fusion PCR and gene targeting in Aspergillus
nidulans. Nature Protocols 1:
3111-3120.
Utermark, J., and P. Karlovsky.
(2008). Genetic transformation of filamentous fungi by Agrobacterium tumefaciens. Protocol
Exchange: 1-17.
Zhou, Y. J., F. Yang,
S. Zhang, H. Tan, and Z. K. Zhao. (2011). Efficient gene disruption in
Saccharomyces cerevisiae using marker cassettes with long homologous arms
prepared by the restriction-free cloning strategy. World J Microbiol Biotechnol 27: 2999-3003.
Return to FGR 60 Table of Contents
FGSC Homepage