A quick RNA mini-prep for Neurospora mycelial cultures
Lindgren, K.M., A. Lichens-Park, J.L. Loros and J.C. Dunlap Dept. of Biochemistry, Dartmouth Medical
School, Hanover, NH 03756.
Most RNA isolation techniques currently in use have been developed for the processing of large
quantities of material. These typically involve multiple phenol extractions (Reinert et al. 1981 Mol. Cell
Biol. 1:829-836) or guanadinium isothio-cyanate/cesium chloride gradients (Chirgwin et al. 1979 Biochem
18:5294-5299) and can be both expensive and time consuming. Often, however, needs arise where
quantitatively smaller amounts of RNA are needed from many different samples, for example, during time
series analyses or when screening transfor-mants for expression of a transformed gene. Under such
circumstances, existing tech-niques are overly time consuming and yield more RNA than is necessary. The
availability of a rapid RNA mini-prep is thus desirable. Such a system has been developed for isola-ting
plant RNA (Nagy et al. 1988 Plant Molecular Biology Manual, B4; ed. Gelvin and Schilperoort, Klewer
Academic Publishing, pp. 1-29), and we have adapted this procedure for use with Neurospora and,
potentially, other filamentous fungi. Below, we describe the use of this procedure with 50 ml mycelial
cultures, although we have used in with equal success with 5 ml cultures without scaling down the amounts
of any reagents.
The method involves the use of a triphenylmethane dye, aurintricarboxylic (ATA), to protect the RNA.
ATA binds irreversibly to RNA and is a potent inhibitor of most nucleic acid binding enzymes (Hallick
et al. 1977 Nucl. Acids Res. 4:3055-3064). Thus, RNA made with procedure cannot be used for in vitro transcription or translation or reverse transcription but works fine for RNA/DNA or RNA/RNA
hybridizations.
To minimize RNase contamination, all glassware is baked at 182°C for at minimum of four hours.
Work with gloved hands. The procedure is as follows:
1. Conidia from slants (grown in 16 x 150 mm test tubes containing 8 ml of solid medium) are
resuspended in 50 ml of Horowitz complete medium (Horowitz 1947 J. Biol. Chem. 171:255-262) and the
cultures grown overnight with shaking at 30°C. A 50 ml culture typically yields enough RNA for 200 gel
lanes (see below), and, as noted, smaller culture volumes may be used.
2. Flat mycelial pads are easier to grind than mycelial balls. Therefore, filter cultures using a Buchner
funnel onto Whatman #44 filter paper. Wrap flat mycelial pads in aluminum foil and freeze in dry ice.
Do not freeze in EtOH/dry ice bath because alcohol might seep through foil. Pads can be stored at -70°C
for at least three weeks.
3. Wash a mortar and pestle thoroughly with warm water and Alconox (Fisher Scientific); cool by filling
with liquid N2. Remove frozen, flat mycelia from foil and add it to the liquid N2 in mortar. Add ~0.5 g
of baked sand and grind mycelial pad to a fine powder. Add more N2 as needed. Mortar and pestle
should be washed after every sample.
4. Working quickly before powder can thaw, pour or spoon ground mycelia into 15 ml round bottom
Sarstedt tube (Sarstedt tubes #60.540, Princeton, NJ) containing 8 ml of E buffer at room temperature.
[E buffer: 50 mM Tris-Cl pH 8.0, 300 mM NaCl, 5 mM EDTA, pH 8.0, 2% SDS; autoclave and add 1 mM
ATA and 14 mM ß-mercaptoethanol. ATA=aurintricarboxylic acid, ammonium salt (Sigma #A0885, St.
Louis, MO)]
5. Thaw the powder in E buffer in 42°C water bath, occasionally shaking, to get SDS into solution. This
should take about 5 minutes.
6. Add 1.1 ml of 3M KCl, invert to mix, keep on ice for 10 min. Solution should form semi-solid,
flocculent mass as K-SDS precipitate forms.
7. Spin at 3000g, 4°C in a fixed angle rotor. Make sure caps are screwed on tightly to prevent tubes from
collapsing.
8. Pass supernatant through 50 micron Miracloth (Calbiochem #475855, La Jolla, CA) in a funnel into
fresh Sarstedt tube.
9. Measure volume of average-sized sample. Add 0.5 vol. 8 M LiCl, mix and stand at 4°C overnight.
10. Spin at 12000g, 4°C, for 15 min in a fixed angle rotor. Thoroughly resuspend pellet in 4 ml sterile
gd (glass distilled) H2O with pasteur pipette.
11. Extract twice with phenol/chloroform/isoamyl alcohol (25:24:1), spinning 12000g 10 min at 4°C in a
fixed angle rotor. Save aqueous (upper) phase; add gd H2O if volume is less than 2 ml.
12. Add 0.1 vol 3M NaOAc pH 6.0, mix and add 2.5 vol EtOH, mix. Place at -20°C overnight or -70°C
for 15 min.
13. Spin 12000g, 10 min, 4°C. Wash pellet with 70% EtOH and drain. Pellet should be a light pink or
white.
14. Resuspend in 0.4 ml sterile gd H2O in a microfuge tube. Precipitate with NaOAc and EtOH as in
step 12.
15. Spin 10-20 min in microfuge. Wash twice with 70% EtOH and dry pellet. Resuspend in 200 µl of
sterile RNase-free gd H2O. Store at -70°C for up to three months. Spectophotometric quantification may
be done at this point.
16. Load 1 µl onto a formaldehyde gel. Electrophorese overnight at 20 volts. Blot onto nitrocellulose.
Probe with DNA fragment of choice. Expose to film.
Yields are typically on the order of 1-2 mg total RNA from an overnight 50 ml culture arising from
an average slant. The number of samples able to be processed using this procedure is limited by the
number of spaces in a centrifuge rotor. We have done as many as 24 samples in one day, and doing
several times this many would be possible. We have observed on ethidium bromide stained gels that the
fluorescence from the RNA deriving from this miniprep is brighter than that seen when the corresponding
amount of standard-prep RNA is used. This may be due to enhanced fluorescence of RNA in the
presence of ATA. However, autoradiography of the blots does not show any RNA degradation products
(Figure 1). This procedure would probably work fine with other methods of tissue disruption. ATA
inhibits many nucleic acid binding proteins, possibly by competing for binding sites (Blumenthal and
Landers 1973. BBRC 55:680-688). Therefore, the most critical factor is getting the RNA in contact with
ATA before nucleases can bind to the nucleic acid and degrade it. Supported by federal grants to J.J.L.
and J.C.D.
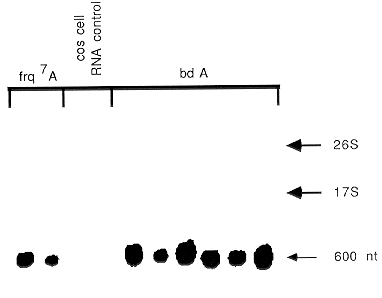
Fig. 1. ATA mini-prep RNA probed with ccg-1 DNA fragment. Total RNA from a series of
transformants into bd A and frq7 A was examined for the presence of the ccg-1 gene transcript (Loros et
al. 1989 Science 243:385-388). Each lane contains 10 µg of RNA (1/200 of the preparation). While the
fluorescence staining of the RNA extended from the 26S to below the 17S RNA bands (not shown), the
hybridization revealed the presence of only a single undegraded transcript in each lane containing
transformant RNA and no hybridization to the monkey cos cell RNA control.
Return to the FGN 37 index
Go to the FGSC main page