Versatile fungal transformation vectors carrying the selectable bar gene of Streptomyces
hygroscopicus
B. Straubinger(1), E. Straubinger(1), S. Wirsel, G. Turgeon and O. Yoder - Department of Plant
Pathology, Cornell University, Ithaca NY 14853. (1)Current address: Consortium f.
Elektrochem. Industrie GmbH, Zielstattstr. 20, D-8000 München 70, Germany
Several selectable genes have been reported for construction of filamentous fungal
transformation vectors. Among the most widely used is the hygB (also known as hph) gene
of E. coli, which is generally useful because the corresponding selective agent (hygromycin
B) is toxic to wild type strains of many fungi and because scoring of transformants is usually
unambiguous. We, and others (Avalos et al. 1989 Curr. Genet. 16:369-372), have found that
the same merits are evident using bialaphos (or phosphinothricin) as a selective agent and
the bar gene (DeBlock et al. 1987 EMBO J. 6:2513-2518), which encodes phosphinothricin
acetyltransferase, as a selectable marker. We report here the construction of three vectors
which carry bar as the selectable gene and have easily exchangeable parts as well as
convenient cloning sites.
The first plasmid (pBP1) was constructed as follows. A 575 bp BamHI fragment
carrying the bar coding region was inserted into the BamHI site of pUC18 (Yanish-Perron
et al. 1985 Gene 33:103). A 631 bp SalI-DdeI fragment carrying the Cochliobolus
heterostrophus Promoter 1 element (Turgeon et al. 1987 Mol. Cell. Biol. 7:3297-3305) was
end-filled, attached to XbaI linkers, and inserted into the pUC18 XbaI site immediately 5'
of the BamHI site, thus creating a Promoter 1::bar transcriptional fusion (Fig. 1). The
second plasmid (pBP1T) was made by inserting a 470 bp AccI fragment (blunt-ended and
attached to EcoRI linkers) from the 3' untranslated region of the C. heterostrophus TRP1
gene (Turgeon et al. 1986 Gene 42:79-88) into the EcoRI site of pBP1, thus providing a
fungal terminator (Fig. 1). The junction regions were sequenced as shown below:
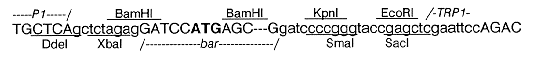
Restriction enzyme sites are underlined or overlined. Linker and vector sequences are in lower case
letters. The 3' end of the Promoter 1 (P1) fragment is shown fused to the XbaI linker. Both ends of the BamHI
fragment carrying the bar gene are shown (internal sequences are omitted); the start codon is in bold type. The
5' end of the TRP1 terminator fragment is shown fused to the EcoRI linker.
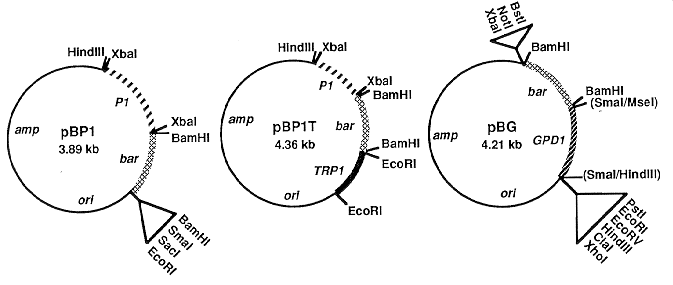
Figure 1. Construction of plasmids pBP1, pBP1T and pBG. Steps in construction are described in the text.
Restriction enzyme sites shown are either unique (for cloning) or points at which the sequences indicated in the
text were inserted. amp = E. coli ampicillin resistance gene; ori = E. coli origin of replication; P1 = C.
heterostrophus Promoter 1; bar = E. coli bialaphos resistance gene coding region; TRP1 = C. heterostrophus
tryptophan biosynthetic gene terminator; GPD1 = C. heterostrophus glyceraldehyde-3-phosphate dehydrogenase
gene promoter.
An important feature of these plasmids is that their relevant parts (promoter, coding
region, terminator) can be readily removed or exchanged with other sequences, simply by
digestion with the appropriate enzyme (XbaI, BamHI or EcoRI) and religation with or
without a substitute fragment attached to the proper linkers. Note that some of the unused
polylinker sites of pUC18 are no longer unique because they are also found in one or more
of the inserts. Remaining cloning sites are EcoRI, SmaI, SacI and HindIII for pBP1 and
HindIII only for pBP1T.
The third plasmid (pBG) was made by inserting the 575 bp BamHI bar fragment into
the BamHI site in the polylinker of the Bluescript vector pIIKS+. A 675 bp HindIII-MseI
fragment bearing the promoter of the C. heterostrophus GPD1 gene (VanWert and Yoder
1992 Curr. Genet. in press), was end-filled and inserted into the SmaI site of pIIKS+, just
5' of the bar gene, thus creating a GPD1 promoter::bar transcriptional fusion (Fig. 1). A
combination of sequencing and restriction enzyme analysis confirmed the junction regions
as shown below.
 Conventions are as described above for pBP1T. Restriction enzyme sites in parentheses are
nonfunctional.
Conventions are as described above for pBP1T. Restriction enzyme sites in parentheses are
nonfunctional.
All three plasmids were used to transform C. heterostrophus, using standard
procedures (Turgeon et al. 1987 Mol. Cell. Biol. 7:3297-3305). The protoplast regeneration
medium for selection of transformants was modified to contain only osmoticum and
Cochliobolus minimal salts (Leach et al. 1982 J. Gen. Microbiol. 128:1719-1729), solidified
with 1% agarose and containing either bialaphos or phosphinothricin at a final concentration
of 50-100 ug/ml. Complex media were avoided since phosphinothricin (a synthetic
compound also known as glufosinate-ammonium, an analog of L-glutamic acid) specifically
inhibits glutamine synthetase. Bialaphos, a naturally-occurring tripeptide consisting of
phosphinothricin and two residues of L-alanine, is toxic to cells after it is converted to
phosphinothricin by endogenous cellular peptidases which remove its L-alanine residues.
The transformation frequency with each of the plasmids was 1-10 fast-growing and
50-500 slow-growing colonies/ug plasmid DNA, comparable to the frequencies obtained
using similar plasmids but with the hygB gene substituted for bar. Integration of either
pBP1 or pBP1T into chromosomal DNA occurred at both Promoter 1 and at ectopic sites.
Single and multiple plasmid copies were observed at either type of site. When
transformants were crossed to wild type, the bar gene segregated as a single mendelian
element, indicating that integration occurred at a single site in each case. pBP1 and pBP1T
were also used to transform Colletotrichum graminicola, using procedures similar to those
described for C. heterostrophus.
Acknowledgements: The bar gene, in plasmid pGSFR1, was obtained from Plant Genetic
Systems N.V., Laboratories Gent, Jozef Plateaustraat 22, B-9000 Gent, Belgium.
Phosphinothricin was from Riedel de Haen AG, Wunstorferstrasse 40, D-3016 Seelze 1,
Germany and bialaphos was from Meiji Seika Kaisha, Ltd., Research Laboratories,
Morooka-Cho, Kohoku-ku, Yokohama 222, Japan. The work was funded by grants from the
U.S. Department of Agriculture and the Cornell Biotechnology Program; B.S. and S.W. were
supported by fellowships from the Deutsche Forschungsgemeinschaft.