The complicating circumstances for this gene are a) increased sensitivity of protoplasts to benomyl as compared to conidia or non-protoplasted fragments of mycelium, and b) decreased viability and growth rate of cells carrying the mutant allele in absence of the selective agent. The donor was a MNNG-induced mutant of P. canescens resistant to 1 ug/ml benomyl which exhibited abnormal growth in the absence of benomyl. Total genomic DNA was isolated, purified by CsCl density gradient ultracentrifugation and partially digested by Sau3A restriction enzyme to obtain an average fragment length of 6-8 kb. The cloning vector pBB8 (construction described in Aleksenko 1994 Curr. Genet. in press, where it is referred to as pCIbb8), contained the AMA1 element, promoting autonomous replication in Aspergillus, and the TrpC gene for initial selection of transformants. The vector was made linear at its unique BamHI site. One hundred milligrams of the vector was mixed with 200 mg of the donor DNA and used in transformation of about 10^8 viable protoplasts of A. nidulans strain yA2 argB2 trpC801. The transformation mixture was plated in 5 ml of stabilized top-agar in standard Petri dishes with minimal medium supplemented with arginine. After 24 hours of regeneration and growth at 37oC, the same amount of top-agar with 4 ug/ml benomyl was overlayed.
After 5 days of incubation, one benomyl-resistant colony was observed. The purified transformant grew on minimal medium supplemented with arginine at benomyl concentrations up to 2 ug/ml. The transformant mycelia were grown separately in liquid minimal medium supplemented with arginine only, and with arginine and tryptophan plus 1 ug/ml benomyl. Plasmid rescue in E. coli was performed as described previously (Gems et al. 1994 Mol. Gen. Genet. in press), essentially with the PEG-DMSO method of transformation (Chung and Miller 1988 Proc. Natl. Acad. Sci. USA 86:2172-2175). Plasmids of the same size and restriction pattern were rescued from both mycelial preparations (designated pABR3; see Fig. 1). They transformed A. nidulans at an efficiency of about 150 colonies per mg (under the conditions described above); however, all the colonies failed to grow after the first or the second passage on benomyl. The stability of the initial transformant was examined by plating conidia on non-selective medium and subsequent replica plating onto media with benomyl or without tryptophan. Of 250 colonies examined, 248 were benS trpC, two were benR trp+, and no colonies of recombinant genotypes were found. These data indicate that the cloned benR allele is linked to the vector marker in an inefficiently replicating plasmid. An attempt to subclone a smaller fragment carrying the benomyl-resistance determinant yielded no transformants. In a P. canescens wild-type strain, where the initial selection for prototrophy is not applicable, neither the initial plasmid nor subclones were able to produce benomyl-resistant colonies.
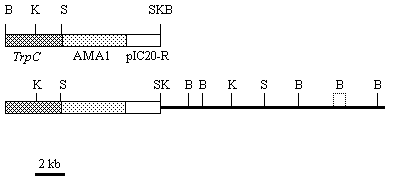
Fig. 1. Linearized restriction maps of the cloning vector pBB8 (top; open and filled-in boxes) and the plasmid pABR3 (bottom), containing the insert of P. canescens genomic DNA (bold line) promoting benomyl resistance to A. nidulans transformants. The sites for restriction enzymes: B, BamHI; K, KpnI; S, SmaI. The BamHI sites to both sides of the insert were damaged by the cloning procedure. The exact position of the second BamHI site from the right cannot be determined by restriction mapping.
The extremely low stability of transformants (less than 1% against 30-60% for control pBB8 transformants) may be due to the large size of the plasmid obtained. Another, and more probable, explanation is that high copy number (up to 30 molecules per cell, as estimated by Gems et al. 1991 Gene 98:61-67) may be essential for phenotypic stability, since it is obvious that plasmid copies segregate extremely irregularly between daughter nuclei. At the same time, large copy number of the mutant benR allele may be lethal for cells. This, rather than organization of the cloned gene as a very long transcription unit, may have caused the failure to subclone a smaller functional fragment, since smaller plasmids may be able to replicate more efficiently and be even more destructive for the host cell. Since the plasmid obtained does not appear suitable for constructing a convenient vector for transformation, I do not intend to continue this study. I am ready to provide all the materials and detailed data to those who are interested in their application.
This work was supported by the Wellcome Trust Foundation.