The catalytic subunit of PP2B from N. crassa was cloned and expressed (Higuchi et al. 1991 J. Biol. Chem. 266:18104-18112). We assumed that PP1 and PP2A were also present in this organisms. In order to test this possibility we assayed phosphatase activities in N. crassa. For the detection of PP1 and PP2A we adapted a method originally described for S. cerevisiae (Cohen et al. 1989 FEBS Lett. 250:601-606). PP2B activity was measured according to Higuchi et al. (1991 J. Biol. Chem. 266:18104-18112).
Neurospora crassa strain RL3-8 (FGSC stock number 2218) was cultured in Vogel's minimal medium for 48 h at 27oC. Two agar slants (one week old) were used to inoculate 400 ml of medium. Mycelia were harvested by filtering through cheesecloth. The mycelial mat was weighed, frozen in liquid nitrogen and stored at -70oC. Homogenization was carried out by disintegrating the mat at the temperature of the liquid nitrogen in mortar with pestle, then by mixing the powder with 4 volumes/weight extraction buffer (50 mM Tris-HCl pH 7.4, 1 mM EDTA, 0.1% vol/vol. beta-mercaptoethanol) containing a mixture of freshly diluted protease inhibitors (5 mM benzamidine, 0.2 mM phenylmethyl sulfonyl fluoride, 1 mM o-phenanthroline). Extraction was completed by mixing the ice cold homogenate at full speed for 1 min in an MSE blender. The extract was filtered through glass wool and was centrifuged at 4oC and 15,000 g for 15 min. The supernatant was used as crude extract in the phosphatase assays. We found that small aliquots (0.1-0.5 ml) of extract can be stored, after being frozen in liquid nitrogen, at -70oC for 7 days without significant loss of phosphatase activity.
PP1 and PP2A were assayed with rabbit muscle phosphorylase a substrate. Phosphorylase a was prepared from phosphorylase b with [gamma-32P]ATP and phosphorylase kinase. The assay mixture contained 5 uM 32P-phosphorylase a dimer and 5 mM caffeine in the extraction buffer in a total volume of 30 ul. The reaction was initiated by the addition of the substrate and was terminated after 10 min incubation at 30oC by the addition of 100 ul 10% TCA. TCA soluble radioactivity was counted by Cherenkov radiation. Maximal specific activity was obtained if the extract was diluted with extraction buffer, so that less than 20% of the total 32P was released from the substrate. To test if the liberated radioactivity was in Pi or small phosphopeptide(s) we converted Pi into phosphomolybdic acid and extracted it with organic solvents (Antoniw and Cohen 1976 Eur. J. Biochem. 68:45-54). More than 95% of the radioactivity was recovered in the organic phase. In addition, 20 mM NaF (a well known phosphatase inhibitor) reduced the activity to 5-8%. These facts demonstrate that the assay measures phosphatase activity and the effect of proteases can be neglected.
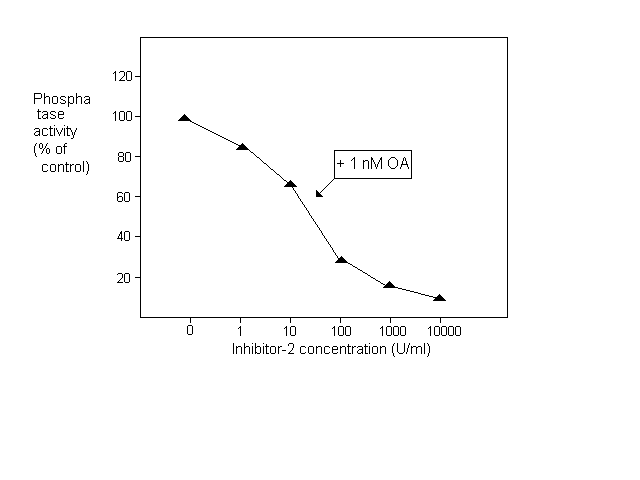
After establishing the assay system we investigated the effect of specific phosphatase inhibitors. One micromolar okadaic acid caused a nearly complete inhibition with the half maximal effect at 10 nM (Fig.1.). Since the IC50 of okadaic acid is 0.1 nM for PP2A and 15- 20 nM for PP1 in mammalian tissue extracts (Cohen et al. 1989 FEBS Lett. 250:596-600) we assumed that most of the phosphorylase phosphatase activity in N. crassa extract was due to PP1. To test this hypothesis we measured the effect of okadaic acid in the presence of an excess of inhibitor-2. As shown in Fig. 1, under these conditions the activity was half maximally inhibited by 0.3 nM okadaic acid and 90% inhibition was observed at 1 nM concentration. The sensitivity of N. crassa PP1 to the rabbit muscle inhibitor-2 was demonstrated in Fig.2. When PP2A was inactivated with 1 nM okadaic acid, 10,000 U/ml inhibitor-2 caused complete inhibition. 50% inhibition was observed at 30 U/ml concentration i.e. around 1 U inhibitor-2 in the 30 ćl assay mixture elicited half maximal inhibition.
Figure 1. Inhibition of phosphorylase phosphatase activity by okadaic acid in the presence and absence of inhibitor-2 (I-2)
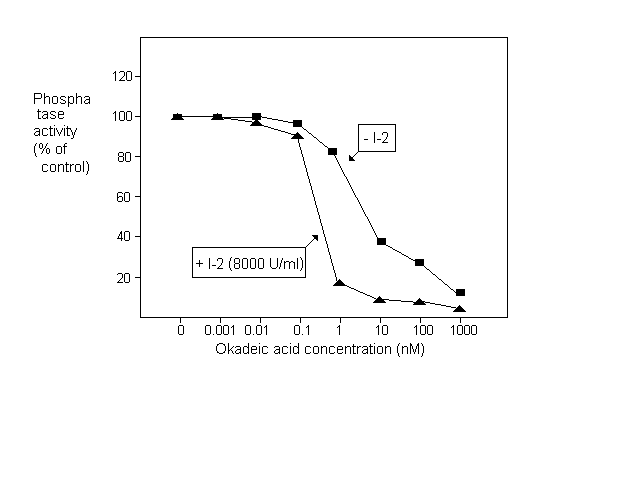
Figure 2. Inhibition of phosphorylase phosphatase activity by inhibitor-2 in the presence of okadaic acid (OA)
Our results indicate that the phosphorylase phosphatase activity inhibited by 1 nM okadaic acid can be attributed to PP2A and the one inhibited by 10,000 U/ml inhibitor-2 belongs to PP1. Protein concentration was measured according to Read and Northcote (1981 Anal. Biochem. 116:53-64). Using these methods we found 4.95 +/- 0.9 mU/mg protein (n=10) of total phosphorylase phosphatase activity in N. crassa mycelial extract, 20% of which was PP2A and 65% PP1. By definition one unit of the enzyme liberated 1 umol Pi in 1 min under the above conditions.
PP2B was assayed with rabbit muscle inhibitor-1. The substrate was phosphorylated with [gamma-32P]ATP and bovine cAMP-dependent protein kinase. The reaction mixture contained 3 uM 32P-inhibitor-1, 40 mM Tris/HCl pH 7.0, 0.4 mM DTT, 0.2 mM CaCl2, 1 mM MnCl2 and 12 uM calmodulin in 30 ul total volume. The reaction was started by the addition of substrate and after 10 min incubation at 30oC it was terminated by the addition of 100 ul 20% TCA and 100 ul 6% BSA. The released radioactivity was counted as above. Using the extraction method, we proved that more than 95% of the TCA soluble radioactivity was due to the presence of 32Pi. About 40% of the total activity was inhibited by 0.5 mM EDTA. The activity stimulated by Ca2+-calmodulin in the presence of Mn2+ was attributed to PP2B, thus the specific activity of PP2B was estimated to be 0.044 +/- 0.01 U/mg protein (n=6) in the extracts. One unit of PP2B liberated 1 nmol Pi in 1 min in the above assay.
The assays described in the present communication are suitable to test protein phosphatase mutants by measuring crude N. crassa extracts, and provide a handle for the purification and characterization of the respective enzymes. The results show considerable similarity between the N. crassa and animal protein phosphatases suggesting that cloning of PP1 and PP2A catalytic subunits could be possible using heterologous cDNA probes.
Acknowledgements. This work was supported by the grants OTKA 6005 and 1501. Sz.B. is a recipient of a fellowship from the Hungarian Academy of Sciences, and the Foundation for the Hungarian Science.