An economical strategy for chromosome walking in the Neurospora crassa
pMOcosX library
Frederick J. Bowring and David E.A. Catcheside - School of Biological Sciences, Flinders
University, GPO Box 2100, Adelaide, SA 5001, Australia.
Chromosome walking using ordered genomic libraries is demanding of both labor and
materials. Such libraries are often stored as a set of 96 well microtiter plates with each well
containing an individual clone. The library is typically prepared for screening by replicating
plates onto ~11 x 7 cm membranes placed on a suitable growth medium. Colonies are lysed
in situ and the cellular material washed off leaving DNA bound to the membranes
(colony blots). To extend the walk, the library is screened by probing the set of colony blots
for homology to sequences close to the terminus identified in the preceding step. Once
clones are identified, they are mapped for one or more restriction enzymes, aligned, and a
sequence extending the walk in the desired direction is selected to initiate the next phase.
The Neurospora crassa genomic library pMOcosX (Orbach 1994 Gene
150:159-162) comprises 50, 96 well microtiter plates. A conventional screening program uses
50 membranes and, due to the practicalities of hybridizing 50 membranes simultaneously,
sometimes several rounds of hybridization. In this note we describe a chromosome walking
strategy in the pMOcosX library that minimizes the scale of screening and also
obviates the need for restriction mapping prior to commencing the next step, which in our
experience is a rate limiting factor. We have used this method to establish contigs of 110
and 240 kb on LG V.
Overview: We made 50 DNA pools, each containing all the clones on one of
the 50 plates, arrayed these on a single 5 x 10 cm membrane and screened this for homology
to probes of relevant sequences. The address of specific clones hybridizing to the probe was
then determined by screening colony blots prepared from the appropriate plate(s). We have
tested the efficacy of this method and present data showing results comparable to screening
a full set of 50 colony blots are achieved. At each step in the walk, the relative positions of
cosmid ends was fixed by end-specific riboprobes transcribed from the T3 and T7 primers
in the vector. The probe transcribed from the cosmid end most remote from the initiation
point of the previous step was used to initiate the next step. We ensured walks were on
track by checking that polymorphisms highlighted by clones had the appropriate linkage
when probed to a panel of progeny from a cross segregating genetic markers on LG V
(Bowring and Catcheside, to be published elsewhere).
Methods: The library was replicated onto 15 cm diameter plates
containing Luria-Bertani medium, ampicillin (50 µg/ml) and solidified with 2% agar. After
incubation at 37°C for 20 h plates were flooded with 10 ml STE (100 mM NaCl, 10
mM Tris, 1 mM EDTA, pH 8.0) and left 30 min. Cells were then easily suspended by brief
agitation. The suspension was transferred to tubes and vortex mixed. DNA was prepared
from 1.5 ml samples by the method of Sambrook et al. (Molecular Cloning, Cold Spring
Harbor 1989). Cells were pelleted in a microfuge, resuspended in 100 µl solution I (50 mM
glucose, 25 mM Tris, 10 mM EDTA, pH 8.0) and mixed with 200 µl solution II (0.2 N
NaOH, 1% SDS). Following the addition of 150 µl solution III (3 M potassium: 5 M
acetate), tubes were vortexed gently in an inverted position for 10 sec and left on ice for 10
min. Cell debris were removed by microfuging at 4°C for 5 min. DNA was precipitated from the supernatant with 2.5 vol ethanol at room temperature for 5 min and pelleted in a microfuge at 4°C for
10 min. Ethanol was removed by aspiration, the pellets were dried briefly in air and
resuspended in 50 µl TE. This provided enough DNA for 10 library screenings. For each
plate pool, the 50 µl DNA preparation was denatured with 50 µl 0.4 M NaOH at
65°C for 15 min and 10 µl immediately spotted onto the same position on each of 10
positively charged nylon membranes (PCNM:Qiabrane plus) in a 10 x 5 array with spots at
1 cm centers. Membranes were baked at 80°C for 30-60 min and used immediately
for hybridization or stored sealed in plastic bags at room temperature. We have identified
positive plates using membranes that have been stored in this fashion for as long as 9
months.
The walk was extended with end-specific riboprobes transcribed from the T3 or T7
RNA polymerase promoters contained in the vector and labelled with 35S-UTP using a Maxi-script kit (Ambion Inc., Texas). Membranes were
prehybridised at 68°C for 1 hr in PE (0.133 M sodium phosphate, pH 6.9, 1 mM
EDTA) + 7% SDS (Reed 1990 Today's Life Science 2:52-60), probed overnight in the same
buffer, washed twice at room temperature for 15 min in 2x SSC/0.1% SDS, twice for 15 min
at 68°C in 0.1x SSC/0.1% SDS, dried and exposed directly to X-ray film
overnight.
Plates containing positive clones were replicated onto PCMN, colonies lysed in
situ and positive clones identified by hybridization. We have found that riboprobes
used in the initial plate pool screening and stored in the interim at -20°C, can be
satisfactorily used to probe colony blots. Thus a clone extending the walk can be located
using as few as two membranes and one labelling reaction.
Efficacy of the plate pool screening method: Using conventional colony blots
of the pMOcosX library, Irelan and Selker (1993 Fungal Genet. Newsl.
40:84) identified six am containing cosmids and M. Lewis four
leu-5 cosmids (personal communication). To model the sensitivity of our plate
pool method, we have probed plate pools with a 924bp BamH1/EcoRV fragment containing
part of the am coding sequence and also a 3.5kb PstI fragment
containing leu-5 coding sequences (Ming-Chow and RajBhandary, 1993 J.
Bacteriol 175:370-379). The probes labelled by random priming with 35S dATP were made
from fragments excised from am and leu-5 plasmids kindly provided by Dr. J.A. Kinsey and
Dr. C. Ming-Chow, respectively. For leu-5, probing our plate pools highlighted all four plates
containing the cosmids identified by Lewis. Irelan and Selker found cosmids G7:2G, G9:10A,
G9:D12, X1:C5, X5:2F and X6:1B each contained am. Our screening showed
each of plates G7, G9, X1 and X6 to contain positive clones but failed to highlight plate X5.
A Southern of cosmids from our copy of the library digested with HindIII and NotI was
probed with DNA from X1:C5. Numerous bands were highlighted in G7:2G and X6:1B but
only vector sequences in X5:2F, suggesting our inability to detect plate X5 with the am
fragment reflects a difference at the X5:2F address between our copy of the pMOcosX
library and that used by Irelan and Selker, rather than a failure of the plate pool detection
protocol.
Extending a walk by mapping end probes. A conventional strategy for
orienting overlapping cosmids is to generate restriction maps with one or more enzymes and
place similar map sections in register. This works well and may give warning of the presence
of non-contiguous DNA. A rapid alternative we have used is to probe dot blots of clones
with riboprobes prepared from all new clones. Riboprobes from cosmids extending the walk
will hybridise only to the clone they were made from. The principles involved are illustrated
in a hypothetical example (Figure 1). This example is a unidirectional walk. For a
bidirectional walk mapping of end probes could be used for extension from the initial clones
since orientation is not immediately relevant. We have ensured our walks are not switched
by jumbled cosmids to another chromosome by checking linkage of RFLPs within clones to
markers in the vicinity of the walk. Once the materials have been prepared, this is a simple
way of ensuring a walk is on the correct chromosome and therefore valuable irrespective
of the walk extension strategy. We have found numerous RFLPs highlighted by LG V
cosmids in laboratory strains. As an example, three out of three cosmids (one near leu-
5, one between am and his-1 and the other a pSV50 cosmid
23:1A containing al-3 and pho-2: Supplement to FGN
40:100) highlighted at least one HindIII RFLP in a pair of strains of
interest to us because we had found them to have RFLPs highlighted by the am containing
cosmid G9:A10. Linkage of the RFLPs in cosmids to other markers can be established
simply by sequentially stripping and reprobing a Southern of genomic DNA from cross
progeny cut with a convenient restriction enzyme. In our case this was HindIII.
Progeny of the Metzenberg mapping crosses (vide Metzenberg and Grotelueschen, 1993
FGN 40:130-138) available from FGSC could be used to ensure walks remain on the linkage
group from which they were initiated.
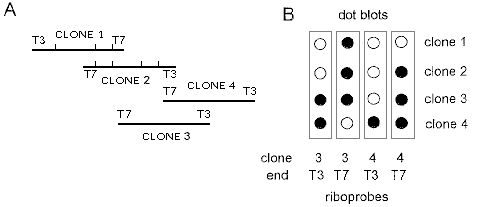
Figure 1A: Clones 1 and 2 were identified by probing the library
with the gene chosen to mark the origin of the walk and mapped with the Stratagene Flash
gene mapping kit. This permitted alignment of the cosmids and determination of which end
of each cosmid,T3 or T7, extends furthest in a specific direction. Extension to the right was
achieved by using a riboprobe from the T3 end of clone 2 to probe the library, identifying
clones 3 and 4.
B: Riboprobes synthesised from the T3 and T7 ends of clones
3 and 4 were used to probe DNA dot blots of all four clones. The T3 riboprobe of clone 4
hybridized only to clone 4 showing it extends furthest in the desired direction and that it is
appropriate to use in the next step of the walk.