A simple dot blot assay to measure hygromycin B
phosphotransferase activity in whole cell extracts of Neurospora
crassa
Michael Freitag and Matthew S. Sachs - Department of Chemistry,
Biochemistry and Molecular Biology, Oregon Graduate Institute of
Science & Technology, Portland OR
Hygromycin B (Hyg) is an aminocyclitol antibiotic with broad
spectrum activity against prokaryotes and eukaryotes (Pettinger et al. 1953 Antibiot. Chemother. 3:1268-
1278). Hyg inhibits protein synthesis by blocking ribosomal
translocation; it prevents polypeptide elongation by interfering
with aminoacyl tRNA recognition and ribosomal A-site occupation
(Cabanas et al. 1978 Euro. J. Biochem.
87:21-27, Hausner et al. 1988 J. Biol.
Chem. 263:13103-13111). Hyg can lead to misreading during
translation in vitro (Davies and Davies 1968 J. Biol. Chem.
243:3312-3316, Gonzales et al. 1978
Biochim. Biophys. Acta 521:459-469, Singh et al. 1979 Nature 277:146-148); however, this effect
was not duplicated in vivo (Bakker 1992 J. Gen. Microbiol.
138:563-569).
Resistance to Hyg is conferred by hygromycin B phosphotransferase
(Hph), first isolated from Streptomyces hygroscopicus
(Leboul and Davies 1982 J. Antibiot. 35:527-528). Hph catalyzes
the phosphorylation of the 4-hydroxyl group on the hyosamine
moiety, thereby inactivating Hyg (Rao et al. 1983 Antimicro. Agents Chemother. 24:689-695).
Plasmid-determined resistance to Hyg had been observed in
Escherichia coli (Rao et al. 1983) and Klebsiella pneumoniae (Gritz and Davies 1983
Gene 25:179-188). Three plasmid-borne genes encoding Hph
were independently isolated and characterized (Kaster et al. 1983
Nucl. Acids Res. 11:6895-6911, Gritz and Davies 1983,
Malpartida et al. 1983 Biochem. Biophys. Res.
Comm. 117:6-12).
Plasmids carrying fusions of a variety of promoters to the
hph gene have been be used to transform eukaryotic cells to
Hyg resistance, including Saccharomyces cerevisiae (Gritz
and Davies 1983, Kaster et al. Curr. Genet. 8:353-358), Aspergillus
nidulans, A. niger (Punt et al.
1987 Gene 56:117-124, Cullen et al.
Gene 57:21-26) and Neurospora crassa (Staben et al. 1989 Fungal Genet. Newsl. 36:79-
81). At least twenty additional species of filamentous fungi have
been successfully transformed to Hyg resistance using two plasmids,
pAN7-1 and pAN8-1, which contain a fusion gene consisting of the A.
nidulans gpd promoter, E. coli hph and the A.
nidulans trpC terminator (van den Hondel and Punt, in: Peberdy
et al. 1991 Applied Molecular
Genetics of Fungi, Cambridge Univ. Press, pp 1-28). The use of Hyg
resistance as a dominant selectable marker in filamentous fungi has
been reviewed (Punt and van den Hondel 1992 Meth. Enzymol. 216:447-
457).
We have used hph as a reporter gene in studies on arginine
(Arg)-specific negative regulation of the N. crassa arg-2
gene. N. crassa host strains were transformed (Selitrennikoff and
Sachs 1991 Fungal Genet. Newsl. 38:90-91) with a plasmid containing
a translational fusion of 5' regulatory arg-2 sequences to hph
(pGV4, Sachs and Freitag, unpublished) or a plasmid containing a
human cytomegalovirus IE94 promoter-driven hph-tk fusion
gene (plasmid tgCMV/HyTK; Lupton et al. 1991
Mol. Cell Biol. 11:3374-3378; tk specifies herpes simplex virus
type 1 thymidine kinase). Single copy ectopic integrants were
selected for further analyses. Transformed strains were resistant
to high levels (>2 mg/ml) of Hyg on minimal growth medium (1X
Vogel's N medium with 2% sucrose and 2% agar). An arg-12s pyr-3 N.
crassa host strain transformed with the arg-2-hph fusion
gene exhibited a phenotype indicating Arg-dependent Hyg resistance;
this strain grows on minimal medium supplemented with uridine (Uri,
0.5 mg/ml) and Hyg (2 mg/ml), but not on minimal medium
supplemented with Arg (0.5 mg/ml), Uri and Hyg.
We desired to quantify Hph activity, but found that the
previously published assay procedures for aminoglycoside-modifying
enzymes were too cumbersome and time-consuming (Haas and Dowding
1975 Meth. Enzymol. 43:611-627). Therefore we adapted a
previously described, simple dot blot assay for measuring Hph
activity from N. crassa whole cell extracts (Duch et al. 1990 Gene 95:285-288,
Sørensen et al. 1992 Gene
112:257-260). Our results show that the hph gene can
be used as a combined selectable dominant marker/reporter gene in
N. crassa strains transformed with plasmids carrying
hph fusion genes.
To analyze Hph activity in N. crassa, aliquots of whole
cell extracts are incubated with of Hyg and [gamma-32P]-
labeled ATP. The samples are filtered through successive layers of
nitrocellulose membrane, P81 phosphocellulose paper and filter
paper. Proteins present in whole cell extracts, some of which are
radioactively labeled by the activity of cellular protein kinases,
are retained on the nitrocellulose membrane, while the weakly
positively charged [gamma-32P]-labeled Hyg passes through the
nitrocellulose and binds to the negatively charged P81
phosphocellulose paper. The filter paper below the
phosphocellulose traps most of the unincorporated [gamma-32P]-labeled
ATP. The amount of [gamma-32P]-labeled Hyg in a dot on
the phosphocellulose paper, quantified by autoradiography followed
by densitometry or by phosphoimager analyses, is used as a measure
of Hph activity.
Experimental. N. crassa conidia (2 x 107 conidia/ml as inoculum) were germinated at 34°C as
shaking cultures (200 rpm on an orbital shaker) for 6.5 h in 125
ml Erlenmeyer flasks containing 30 ml of 1X Vogel's minimal medium
with 2% sucrose, supplemented with 0.5 mg/ml Uri (U) or 0.5 mg/ml
Arg and 0.5 mg/ml Uri (R), as indicated in Figure 1. The cultures
were harvested through Millipore filter assemblies onto Whatman
filter paper (#1). In the cold room, mycelial pads (ca. 0.5 g wet
weight) were added to 0.8 g of acid-washed glass beads (0.5 mm) in
2 ml Sarstedt screw cap tubes containing 1 ml of breaking buffer
(20 mM HEPES-OH, pH 7.9, 100 mM KCl, 2 mM EDTA, 10 mM DTT, 20%
glycerol; Sachs and Ebbole 1990 Fungal Genet. Newslet. 36:35-37).
Tubes were filled completely with breaking buffer and cells broken
by bead-beating in a Mini Beadbeater (Biospec, Bartlesville, OK)
for two 1 min cycles,
interrupted by a 1 min rest on ice. Whole cell extracts were
clarified by centrifugation at 4 C for 10 min at
16,000x g. Extracts were transferred to fresh Eppendorf
tubes and either used immediately or quick frozen in liquid
nitrogen and stored at -80 C. In our experience, extracts
retained Hph activity levels comparable to fresh extracts after 1
year of storage at -80 C.
Serial dilutions of 5, 2.5, 1.25 and 0.63 ug of total protein
(determined by Bradford assay with BSA as standard) from whole cell
extracts in a 10 ul total volume were added to wells of non-sterile
96-well microtiter dishes. The reactions were started by the
addition of 50 ul of reaction buffer (13.4 mM Tris-maleate, pH 7.1,
8.4 mM MgCl2, 80 mM NH4Cl, 60 uM
hygromycin B, 15 uM ATP and 25 uCi/ml of [gamma-32P]-ATP) and incubated
at room temperature for 1 hr. During this time, filter paper
(Micro Filtration Systems, #1514A46X57CM), phosphocellulose paper
(P81 cation exchanger, Whatman #3698 915) and nitrocellulose
membrane (Nitrobind, 0.22 micron, MSI #EP2HY00010) were placed in
this order onto a Biorad BioDot filtration apparatus (two
successive vacuum traps were attached to the apparatus to trap
radioactive efflux). The wells were sealed by application of a
light vacuum and washed with 150 ul water.
The completed reactions were mixed with 150 ul water and heat-
inactivated for 10 min at 70 C. Reactions were loaded into
wells of the Biorad BioDot filtration apparatus and filtered by
immediate application of a light vacuum. Wells were washed three
times with 250 ul water. The apparatus was disassembled and
immediately rinsed until no counts were detected by Geiger counter
and wipe tests. Both nitrocellulose membrane and phosphocellulose
paper were washed three times with water (15 min per wash) at 65 C,
air-dried and exposed to Kodak XAR-5 film (12 h to 3 days with an
intensifying screen at -80 C) or to phosphorimager storage
plates. The filter paper, on which most of the unincorporated
label was detected, was discarded. Amounts of radioactivity in
dots on the phosphocellulose paper were determined with a Molecular
Dynamics Phosphorimager system.
Results and Discussion. As expected, Hph activity was
not detected in the N. crassa wild type strain 74A-
OR23-1VA), while strains transformed with hph-containing
plasmids exhibited readily detectable Hph activity (Figure 1). A
wild type strain transformed with plasmid tgCMV/HyTK showed Hph
activity that was independent of the presence of Arg in the growth
medium (Figure 1), as anticipated. In contrast, an N. crassa arg-
12s pyr-3 double mutant transformed with a
construct containing N. crassa arg-2 5' regulatory sequences
fused to hph showed Arg-specific regulation (Figure 1). The
magnitude of regulation, approximately 3-fold, is similar to that
due to regulation of the arg-2 gene in a wild type strain
(Davis and Ristow 1987 J. Biol. Chem. 262:7109-7117).
Hph activity was proportional to the amount of total protein
assayed (data not shown). We found that stopping reactions by
incubation at 70 C for 10 min and processing the reactions
quickly by applying a light vacuum improved the reliability of the
assay when compared with the previously published procedure
(Sørensen et al. 1992), in
which reactions were not stopped and in which reactions were
filtered by gravity flow alone. In our experience, filtering by
gravity flow leads to weaker signals.
In summary, we show that the Hph activity dot assay first
described by Sørensen et al. can be adapted for use with filamentous fungi. Extracts
from positive and negative control strains should be used in each
experiment to allow comparisons between assays performed on
different days. This requirement is easily fulfilled because Hph
activity in extracts appears to be stable for at least one year
when extracts are stored at -80 C. By using the assay
described here, the hph gene can be used in filamentous
fungi not only as a selectable dominant marker, but also as a
reporter gene.
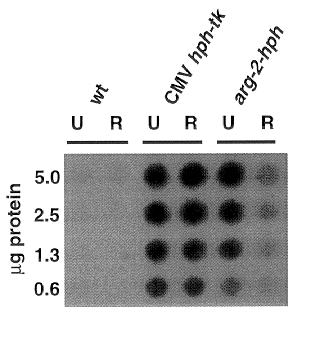
Figure 1: Hygromycin B phosphotransferase dot blot assay on
whole cell extracts of N. crassa. Conidia from wild
type (wt) and hph-containing strains were germinated for 6.5
hrs in minimal medium containing 0.5 mg/ml uridine (U) or minimal
medium containing 0.5 mg/ml arginine and 0.5 mg/ml uridine (R).
Hph activity assays were performed using the amounts of total
protein indicated as described in the text.