Utilization of the Aspergillus nidulans pyrG
gene as a selectable marker for transformation and electroporation of
Neurospora crassa
Gloria E. Turner1, Tommy J. Jimenez1, Suhn Kee Chae1,2,
Rudeina A. Baasiri3,4 and Katherine A. Borkovich3 –
1Department of Chemistry and Biochemistry,
University of California, Los Angeles, CA 90095 and 3Department of Microbiology
and Molecular Genetics, University of Texas Medical School, Houston, TX 77030
Current address: 2Pai Chai University, 439-6 Doma-Dong, Seo-Gu, Tae Jeon,
Korea and 4Department of Cell Biology, Baylor College of Medicine, Houston, TX
77030
We report the complementation of the Neurospora crassa pyr-4 mutation
with the Aspergillus nidulans pyrG gene. The use of the pyrG
gene as a selectable marker for N. crassa transformation offers several
advantages: a non toxic nutritional selection, the ability to put single-copy
transformants through a sexual cross without the occurrence of RIP
(Repeat-Induced Point mutation) by alleles of the pyr-4 gene, and a
relatively-high electroporation efficiency. These traits were exploited for
targeted gene replacement in N. crassa. In addition, the high
electroporation efficiency of pyrG achieved using supercoiled circular
DNA renders it an ideal marker for random insertional mutagenesis studies in
N. crassa.
The A. nidulans pyrG (Oakley et al., 1987 Gene
61:385-399) and N. crassa pyr-4 (Buxton and Radford 1983 Mol.
Gen. Genet. 190:403-405) genes encode orotidine 5'-phosphate
carboxylase, the terminal enzyme in uridine 5'-phosphate biosynthesis. N.
crassa pyr-4 can complement the pyrG89 mutation in A.
nidulans (Waring et al., Gene 79:119-130); however, it was
not known if the A. nidulans pyrG would complement the pyr-4
mutation. The inability to obtain non-leaky mutants at the pyr-4 locus
hampered this testing. We now have available a very tight pyr-4
mutation in both mating types (FGSC 4030 & 4031) that allowed us to test
for complementation of the A. nidulans gene in N. crassa. These
mutants require uridine supplementation for growth.
Southern analysis under high-stringency conditions demonstrated that the
pyrG DNA did not recognize any N. crassa genomic sequences in a
wild type strain. This result is consistent with the very short regions of
identity (maximum of 8 bp in a stretch; 46% identity overall) and numerous gaps
observed after alignment of the DNA sequences of pyrG and pyr-4
(corresponding to the amino acid coding region; data not shown). Taken
together, these observations suggested that pyrG in single copy would
not be subjected to RIP mutation by the presence of a pyr-4 allele
during a sexual cross in N. crassa.
We transformed 7-day old pyr-4 A conidia with supercoiled ppyrG plasmid
DNA using the Vollmer and Yanofsky protocol (1986 Proc. Natl. Acad. Sci. USA
34:573-577). The ppyrG plasmid consists of an EcoRI-NdeI
fragment from pJR15 cloned into pUC19 (Gregory May, personal communication;
Figure 1). The N. crassa pyr-4 gene carried on the pRG1 plasmid (Waring
et al., 1989 Gene 79:119-130; generously provided by Gregory May)
was used as a positive control. Transformants were selected for growth on
minimal medium. The low transformation frequency obtained for both ppyrG and
pRG1 (8 transformants/ ug DNA) was attributed to a 30 minute spheroplasting
step; in subsequent experiments, the spheroplasting time was reduced and the
transformation frequency improved ten-fold.
The original transformation yielded stable transformants for both pyrG
and pyr-4 genes. The transformants were purified to homokaryons by
repeated plating of single conidial isolates. Characterization of homokaryons
included Southern analysis and examination of progeny from crosses to strains
73 a and pyr-4 a. Southern analysis was performed on nine
pyrG transformants and two pyr-4 transformants. We observed
three types of integration events for pyrG transformants; 1) single
ectopic integration (four transformants); 2) multiple ectopic integration (one
transformant); and 3) integration of tandem repeats (three transformants). One
transformant had no detectable pyrG DNA. Of the two pyr-4
transformants, one contained a homologous integration and the other an ectopic
integration event.
All single ectopic integration pyrG transformants produced pyrG+
progeny, indicating that pyrG was not mutated by RIP in N.
crassa. When using the pyr-4 mutant as a female it was necessary to
supplement synthetic crossing medium with 20 mg/ml uridine (Perkins et
al., 1982 Microbiol. Rev. 46:426-570).
To test the utility of the A. nidulans pyrG gene as a selectable marker
for electroporation (Vann, 1995 Fungal Genet. Newsl. 42A:53; Ivey et
al., 1996 Mol. Biol. Cell 7:1283-1297), we electroporated 8-day old
conidia from N. crassa strains pyr-4A and pyr-4a with
ppyrG. Briefly, 40 ul of a solution containing 2.5 x 109 conidia/ml in 1 M
sorbitol (108 conidia total) was mixed with 10 ul of supercoiled ppyrG plasmid
(2 ug) in an electroporation cuvette. Electroporation coniditions were 25 mA,
25 W, 1500 V, 200 ohms and 71 F. After electroporation, 960 ul of 1M sorbitol
was added to the electroporation cuvette. A substantial number of
electroporants was obtained after plating only 5-10 ul of the 1 ml
electroporation solution, with an efficiency of 3400 electroporants/108 conidia
or 1700 electroporants/ ug DNA.
Having determined that pyrG was an efficient marker for
electroporation, we next used this gene as a selectable marker for construction
of a targeted gene replacement mutation in N. crassa (Baasiri et
al., submitted). The gene subjected to mutation was N. crassa
gna-2, which encodes a heterotrimeric G[[alpha]] subunit protein. The
deletion construct (pRAB11) was made by replacing a StuI-NcoI
fragment containing most of the amino acid coding region of gna-2 with
the 2.3 kb EcoRI-NdeI fragment from ppyrG. The construct
contained 2.4 kb and 1.26 kb of 5' and 3' flanking N. crassa gna-2 DNA,
respectively.
Electroporation was performed using 2 ug linearized (to increase the rate of
double crossover events) pRAB11 and 10-day old pyr-4 a or pyr-4 A
conidia. Approximately 450 electroporants were obtained/ ug linearized pRAB11
DNA. Genomic DNA was extracted from 47 pyr-4 a and 37 pyr-4 A
electroporants. Southern analysis revealed that 2 of the pyr-4 a and 3
of the pyr-4 A electroporants possessed a band corresponding to targeted
integration of the deletion construct (and no other ectopic copies of the
electroporation construct), yielding a homologous recombination frequency of
5/84 or 6%. This frequency is similar to that obtained in making gene
replacement mutations in other N. crassa genes using a comparable amount
of homologous flanking DNA (Aronson et al., 1994 Mol. Gen. Genet.
242:490-494; Ivey et al., 1996 Mol. Biol. Cell
7:1283-1297). The five electroporants with the gene deletion were all
heterokaryons. To isolate homokaryotic mutants, each heterokaryotic
electroporant was used as a male in a cross with a N. crassa pyr-4
female of opposite mating type. All ascospore progeny which germinated and
grew into colonies on minimal medium were homokaryons for the gene deletion as
assessed by Southern analysis. The GenBank accession number for the A.
nidulans pyrG gene is M19132. The ppyrG plasmid and pyr-4 mutant
strains used in these studies are available from the Fungal Genetics Stock
Center.
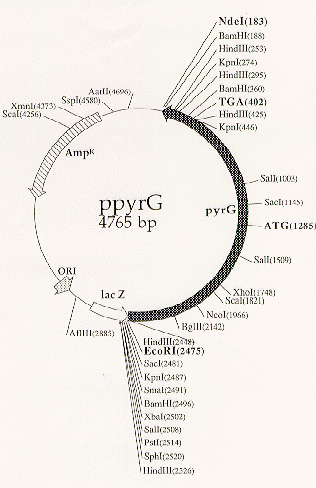
Figure 1. Map of the ppyrG plasmid.
The A. nidulans pyrG gene present on a 2.29 kb EcoRI-NdeI
fragment from pJR15 was subcloned into the same sites in pUC19 to yield ppyrG
(Gregory May, personal communication). All sites for the indicated restriction
enzymes are shown. The bold ATG and TGA denote the pyrG translational
start and stop codons, respectively.
Return to the FGN 44 Table of Contents
Return to the FGSC home page