Rapid procedure for the extraction of DNA from fungal
spores and mycelia.
John J. Weiland- USDA-ARS, Northern Crop Science Laboratory, Sugarbeet and Potato
Research Unit Fargo, N.D. 58105-5677 USA
A method is described for the reliable preparation of DNA from fungal spores
and mycelia and from plant tissues. A number of fungal and plant species were
used in the study to indicate the generality of the method. The DNA prepared
by this protocol was digested by restriction endonucleases and served as
template using standard polymerase chain reaction conditions.
Numerous reports have described procedures for the extraction and purification
of fungal DNA. Many of these are modifications of the CTAB method originally
developed for plant tissue extraction (Saghai-Maroof, et. al. 1984 Proc.
Natl. Acad. Sci. USA 81:8014-8018) or employ direct sample extraction with
organic solvent as the principal means of denaturing and eliminating contaminating
protein (Blin and Stafford 1976 Nucl. Acids. Res. 3:2303-2308).
Whereas the CTAB method is considered superior at removing unwanted carbohydrate from
DNA preparations, procedures that use organic solvents directly in the extraction
buffer often can be performed more rapidly. Despite the range of techniques
available for the preparation of fungal DNA, some fungal mycelia and most fungal
spore samples remain intractable to extraction by these procedures.
The method outlined below was designed to produce a rapid, inexpensive and
reliable procedure for the extraction of total nucleic acids from plant and
fungal tissue, including fungal spores. White quartz sand (-50 + 70 mesh; Sigma Co. product
# S-9887) is used as a pulverizing agent in the protocol, obviating the need for
freezing the sample with liquid nitrogen or for germinating spores prior to DNA
extraction. The method is similar to that described by Chow and Kafer (Fungal
Genet. Newsl. 40:25-27) but is applicable to a wider range of tissue
types. Fungal species tested here include the rust fungi Puccinia
graminis (wheat stem rust), Uromyces appendiculatus (bean rust),
Puccinia helianthi (sunflower rust), and the mildews Plasmopara
halstedii (sunflower downy mildew) and Erysiphe graminis (barley
powdery mildew). Other fungi include the basidiomycete Rhizoctonia
solani, the ascomycete Pyrenophora teres, the deuteromycetes
Phoma betae and Cercospora beticola, and the oomycetes
Aphanomyces cochlioides, and Pythium ultimum. Plant species
included in the study were Beta vulgaris, Solanum tuberosum,
Helianthus annua, and Hordeum vulgare. Recipes for the
phenol/CHCl3/isoamyl alcohol (25:24:1) mixture and the electrophoresis buffers
can be found in Maniatis, et al. (1982, “Molecular Cloning- A laboratory
manual” Cold Spring Harbor Press)
Fresh plant leaf material was weighed directly (for grass species) or after
removal of the petiole and midrib sections (for dicot species). Fungal mycelia
were either scraped directly from the surface of an agar culture or were
trapped on Miracloth (Calbiochem) by filtration of a liquid culture. The
Miracloth was placed mycelia side up onto a large absorbent paper towel to wick
away as much liquid as possible. The final mycelial mat was weighed before
placing it into a mortar. Dry rust and mildew spores were weighed and added
directly to the mortar. NOTE: Doubling of the liquid volumes and mass amounts
of sand is advised as a starting point for application of the procedure to dry
rust and mildew spores.
White quartz sand was added to the tissue at a ratio of 3 g of sand per gram
of tissue. Sufficient extraction buffer [100 mM Tris- HCl (pH 8.0), 20 mM Na2 EDTA,
0.5 M NaCl and 1% sodium dodecylsulfate] was added to
the mortar so that the sand/mycelia mixture became saturated, but no excess
liquid was present (~0.4 ml of buffer per gram of sand). A mixture of buffer
saturated phenol/CHCl3/isoamyl alcohol was added at a ratio of 0.5 ml per gram
of tissue.
The mixture was ground vigorously for ~30 sec with a pestle to form a thick
paste. Two milliliters of extraction buffer and 1 ml of buffered phenol/CHCl3/
isoamyl alcohol per 0.5 g of starting tissue were then added and the solution
was mixed thoroughly. A 1 ml plastic micro-pipette
tip was cut with scissors about 1 cm from the tip and the mixture was
transferred into several microfuge tubes using this tip and a micropipettor.
The microfuge tubes were capped and centrifuged at 16,000 x g for 5 min at room
temperature; tissue debris and the sand pelleted to the bottom of the tube.
The aqueous phase was transferred to a new tube and was mixed with 0.6 vol of
isopropanol. (If needed, the sample can be extracted with phenol/CHCl3/ isoamyl
alcohol one more time followed by centrifugation as above before precipitation
with isopropanol.) Samples were incubated at room temperature for 10 min and
centrifuged at 4 o C for 15 min at 16,000 x g to recover the precipitate. After
rinsing the pellets with 95% EtOH, the pellets were allowed to air dry briefly
and were subsequently resuspended in 340 ul of TE (10 mM Tris-HCl,
1 mM Na2EDTA, pH 8.0) containing ribonuclease A at 20 ug/ml. Samples were
incubated at 37 o C for 30 min were then extracted with 0.3 ml of phenol/CHCl3/
isoamyl alcohol. The aqueous phase (~300 ul) was transferred to a new tube and
1/2 volume of 7.5 M ammonium acetate and 2.5 vol of EtOH were added and mixed,
and the samples were incubated for 30 min at -20 o C. The samples were then
centrifuged at 4 o C for 15 min at 16,000 x g. The pelleted DNA was rinsed with
95% EtOH, air dried and resuspended in 100 ul of TE. Quality and quantity of
the DNA obtained were determined by subjecting a portion of the preparation to
agarose gel electrophoresis and UV spectrophotometry, respectively. Yields
varied from ~10 ug to 50 ug of DNA recovered from 1 g of plant tissue to ~100
ug of DNA obtained from 1 g of rust or mildew spores. A fraction of the DNA
preparations were subjected to gel electrophoresis before (Figure 1) and after
(Figure 2) digestion with restriction endonucleases. A subset of the samples
was also tested as template in polymerase chain reaction experiments (Figure 3).
The method described is reliable, inexpensive and efficient for extracting DNA
from fungal and plant samples. With the inclusion of diatomaceous earth
(bentonite) in the extraction buffer, high quality RNA also can be prepared by
the method (not shown). The protocol adds to the repertoire of nucleic acid
preparations that can be applied to diverse tissue sources.
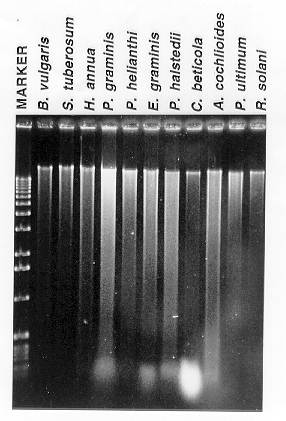
Figure 1. Agarose gel electrophoresis of a subset of the DNA preparations. The
lanes are labelled according to the tissue source. After spectrophotometric
quantification, samples were adjusted to a DNA concentration of ~50 ug/ml and
10 ul of each sample was loaded onto a 1% agarose gel (1X Tris- acetate
electrophoresis buffer). One- half microgram of the 1 kb ladder(BRL- Life
Sciences) was loaded into the "Marker" lane. The gel was stained with
ethidium bromide before photographing with Polaroid 667 film.

Figure 2. Restriction enzyme digestion of DNA prepared by the described method.
Approximately 1 ug of DNA from Puccinia graminis, Rhizoctonia
solani, and Helianthus annua was digested with the restriction
endonucleases BamHI (six base recognition site, GC-rich),
HindIII (six base recognition site; AT- rich),
and HaeIII (four base recognition site). Incubation of the DNA prepared
from Helianthus annua in restriction enzyme buffer in the absence of
added endonuclease (-) indicates the lack of endogenous deoxyribonucleases
contaminating the preparations. The 1 kb ladder is also shown (Marker).
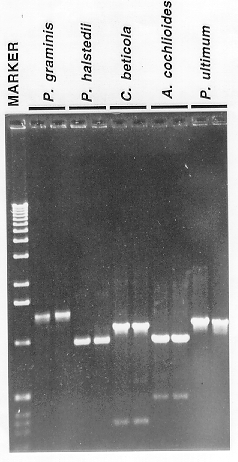
Figure 3. Polymerase chain reaction (PCR) amplification of DNA using fungal DNA
prepared by the described method as template. Reactions were performed in
duplicate using genomic genomic DNA of Puccinia graminis, Plasmopara
halstedii, Cercospora beticola, Aphanomyces cochlioides, and
Pythium ultimum. Opposing oligonucleotide primers were designed based on
consensus sequences derived from the comparison of several fungal actin genes.
Each PCR reaction had a volume of 50 ul and contained 50 mM KCl, 10 mM TRIS-HCl
(pH 9.0), 0.1% TRITON- X100,1.5 mM MgCl2, 0.1 mM each d(G,A,T,C)TP,
50 ng each of the oligonucleotide primers, 10 ng of fungal DNA template and
2 U of Taq DNA polymerase (Promega). Samples were overlayed with mineral oil
and were incubated in an MJR PTC- 100V thermocycler (94o C,
1 min; 55o C, 1 min; 72o C, 3 min : 40 cycles).
A portion (7 ul) of each reaction was electrophoresed in a 1% agarose gel
(1X Tris- borate buffer) in parallel with the 1 kb ladder (Marker).
Return to the FGN 44 Table of Contents
Return to the FGSC home page