Split-Marker Recombination for Efficient Targeted Deletion of Fungal Genes
Natalie L. Catlett1, Bee-Na Lee1, O. C. Yoder1, and B. Gillian Turgeon2,3 1Torrey Mesa Research Institute, Syngenta Research and Technology, 3115 Merryfield Row, San Diego, CA 9212. 2Department of Plant Pathology, Cornell University, Ithaca, NY, 14853
3This work was done while BGT was at TMRI on a leave of absence from Cornell University
Fungal Genet. Newsl. 50:9-11
A commonly used method for fungal gene deletion is introduction of linear DNA consisting of a selectable marker gene flanked on both sides by short stretches of DNA that target a gene of interest (Wirsel et al 1996 Curr. Genet 29:241-249). Gene deletion in Cochliobolus heterostrophus and Gibberella zeae occurs efficiently with this approach. To facilitate deletion construct synthesis, we have applied the "split-marker” deletion strategy previously developed for Saccharomyces cerevisiae (Fairhead et al. 1996 Yeast 12:1439-57; Fairhead et al. 1998 Gene 223:33-46). Here, we describe both fusion PCR-based and plasmid-based deletion methods using this strategy with PEG-mediated protoplast transformation (Turgeon et al, 1985 Mol. Gen. Genet. 201:450-453). These methods are predicted to work well with any transformable fungus that undergoes homologous recombination between chromosomal and introduced DNA sequences.
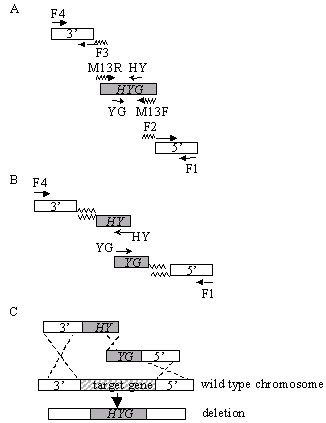
For the split-marker deletion method, two constructs are required per transformation, each containing a flank of the target gene and roughly two thirds of a selectable marker cassette. Homologous recombination between the overlapping regions of the selectable marker gene and between the flank regions and their genome counterparts results in a targeted gene deletion and replacement with an intact marker gene (Fig.1C).
PCR fusion method
The PCR-based strategy eliminates subcloning of the target sequences, and requires only two rounds of PCR reactions. Four universal/selectable marker primers and four gene-specific primers are required for each deletion (Table I).
In PCR round 1 (Fig. 1A), the flanks and the selectable marker are amplified. Primers F1 and F2 amplify the 5' flank; F3 and F4 amplify the 3' flank. The overlapping marker fragments "HY” and "YG” of the hygromycin phosphotransferase cassette (HYG) are amplified from pUCATPH (Lu et al. 1994 PNAS 91:12649-53) using M13R/HY and M13F/YG primers, respectively (Table I). The 5’ extensions for primers F2 and F3, facilitating fusion of the flanks and the marker sequences, are complementary to the M13F and M13R primer sequences, respectively.
Design of the primers to fuse to a standard vector sequence enables their reuse in making deletion constructs with different resistance markers (Amberg et al. 1995 Yeast 11:1275-1280). Moreover, the M13 primer sequences work well for fusion PCR. Stock preparations of the selectable marker cassettes can be made and used repeatedly. For each flank, we normally amplify 250-500 bp; larger flanks may improve transformation efficiency in some fungi. Products are purified using QIAquick PCR purification kit (Qiagen Inc., Valencia, CA) to remove excess primers.
In PCR round 2 (Fig. 1B), each flank from round 1 is fused to the marker through PCR splicing by overlap extension (Ho et al. 1989 Gene 77:51-9). For the 5' construct, templates are the M13F/YG marker fragment ("YG”) and F1/F2 flank from round 1, primers are F1 and YG. For the 3' construct, templates are the M13R/HY marker fragment ("HY”) and F3/F4 flank; primers are HY and F4. Alternatively, we have used the entire M13R/M13F amplified HYG fragment from pUCATPH as a template for both flank fusion reactions.
Standard PCR conditions are used for both rounds of PCR amplification. We have used either Taq (Qiagen) or Expand (Roche Diagnostics Corporation, Indianapolis, IN) polymerases, following the manufacturers’guidelines for PCR conditions. For round 2 fusion PCR, we generally use 1-2 μl of the purified round 1 products as template. Template concentration does not seem critical. Reducing the primer concentration to 50 nM for round 2 reactions may give cleaner results. A 50 μl PCR reaction generally yields more than sufficient DNA for transformation of C. heterostrophus and G. zeae (1-2 μg DNA for each of the 5’ and 3’ constructs). We usually concentrate the second round PCR products prior to protoplast transformation using MinElute PCR purification kit (Qiagen) or concentrate both 5’ and 3’ constructs together using the QIAquick PCR purification kit (Qiagen).
We have deleted numerous genes in C. heterostrophus and one in G. zeae with high efficiency using this method (see Table II).
Plasmid-based method
This strategy allows incorporation of longer target gene flanks into deletion constructs. Also, use of plasmids avoids introduction of unknown PCR-induced mutations, as the flank insert can be sequenced prior to use in transformation, if desired. Each of the two plasmids required for a deletion is made through one-step cloning. Flanks are PCR-amplified using primers that incorporate appropriate restriction sites, then subcloned into a vector plasmid containing either the "HY” or the "YG” portion of hygB (Fig. 2). Each plasmid is digested to release fragments containing the flank and partial marker gene. The unpurified DNA mixture can be directly transformed into protoplasts after restriction enzymes are inactivated or removed.
We have used the plasmid method to delete two C. heterostrophus genes: the ortholog of S. cerevisiae ADE2 (phosphoribosylaminoimidazole carboxylase) and a histidine kinase,HHK12 (Table III, Catlett et al, in preparation). To make the ChHHK12 deletion constructs, the 5' flank XbaI-SpeI (not recommended, compatible ends) was subcloned into pYG and the 3' flank BamHI-XhoI into pHY. Then, the deletion constructs were excised from pYG with XbaI and KpnI and from pHY with SacI and KpnI. Using approximately 1 μg total digested DNA from each plasmid, four ChHHK12 deletion transformants were obtained. Two of these were tested by PCR and confirmed as correctly targeted integrations.
We have adapted the yeast "split-marker” deletion procedure for filamentous ascomycetes, using C. heterostrophus and G. zeae as subjects. This method allows for rapid assembly of deletion constructs and efficient targeted integration of these constructs via protoplast transformation (and likely with other transformation methods, although none has been tested by us). Moreover, the frequency of correctly targeted deletion constructs in fungal systems with less-efficient homologous recombination than C. heterostrophus or G. zeae is potentially increased because only transformants in which the two overlapping marker fragments have successfully recombined will grow in selective medium. Note that our analyses did not formally rule out the possibility that individual fragments integrated at ectopic locations in the genome. This is not a concern with C. heterostrophus, since transforming DNA generally integrates at a single site. Should this be a concern, Southern blots could be done. An additional advantage of this method is the potential for "mix and match” of 5’ and 3’ constructs allowing deletion scanning of a region (Fairhead et al.1996). The procedure can be readily scaled to 96-well format allowing for the assembly of numerous constructs in parallel.
ACKNOWLEDGEMENTS
We would like to thank Scott Baker, Kay Chea, and Jason Hwang, early adaptors of this technique, for advice and encouragement, and David Chou and Kathryn Bushley for technical help with Table II.
Table I. Sample primers for deletion constructs using PCR-fusion method.
M13F |
M13 forward |
5’ CGCCAGGGTTTTCCCAGTCACGAC 3’ |
M13R |
M13 reverse |
5’ AGCGGATAACAATTTCACACAGGA 3’ |
HY |
NLC37 |
5' GGATGCCTCCGCTCGAAGTA 3' |
YG |
NLC38 |
5' CGTTGCAAGACCTGCCTGAA 3' |
F1 |
5’ flank outer |
5’ ATATAACCCTCCGGCCATC 3’ |
F2 |
5’ flank inner |
5’ gtcgtgactgggaaaaccctggcg-AAGGCAAAGTCGGACTTGT 3’ |
F3 |
3’ flank inner |
5’ tcctgtgtgaaattgttatccgct-CGCCTCTTGTCGAGGAGCTA 3’ |
F4 |
3’ flank outer |
5’ AGAATTGCAGCGCCTTCCTA 3’ |
Primers F1, F2, F3, and F4 are gene specific primers designed for the deletion of the C. heterostrophus REC1 gene (see Table II). For F2 and F3 primers, lowercase bold portions are complementary to M13F and M13R sequences, respectively.
Table II. Integration efficiency for selected gene deletions.
gene |
5’ flank |
3’ flank |
total |
correct |
ectopic |
inconclusive |
ChREC11 |
309 bp |
285 bp |
5 |
5 |
0 |
0 |
ChSKN71 |
389 bp |
384 bp |
8 |
8 |
0 |
0 |
ChNPS72 |
546 bp |
514 bp |
16 |
8 |
0 |
8 |
ChNPS92 |
471 bp |
495 bp |
16 |
16 |
0 |
0 |
ChNPS112 |
524 bp |
514 bp |
8 |
8 |
0 |
0 |
GzNTF13 |
761 bp |
420 bp |
12 |
7 |
2 |
3 |
Integrations were assessed using primers outside of the deletion constructs together and in combination with the HY (NLC37) and YG (NLC38) primers. Inconclusive transformants reflect observation of PCR products that could not be clearly interpreted as consistent with either homologous or ectopic integration events. Ectopic events were determined by the absence of PCR products confirming integration and presence of the WT length PCR product obtained by using primers outside of the deletion construct.
Ch = Cochliobolus heterostrophus. Gz = Gibberella zeae.
1Genes described in Catlett et al., (in preparation).
2Gene deletions described in Lee et al., (in preparation).
3Turgeon, unpublished
Table III. Sample primers for plasmid deletion constructs.
NLC28 |
HHK12-1-XbaI |
5’ gcctctagaCCCAAGAGAAAGCTGCCAACGAG 3’ |
NLC29 |
HHK12-2-SpeI |
5’ gccactagTCACCGCGAGGAACCAAAGATAG 3’ |
NLC30 |
HHK12-3-BamHI |
5’ gccggatccATGGCGAGCCAGGTCCAGGTG 3’ |
NLC31 |
HHK12-4-XhoI |
5’ gccctcgagGCTACTTTCTGAAGCGACGAC 3’ |
Primers NLC28 and NLC29 amplify the 5’ flank of ChHHK12. Primers NLC30 and NLC31 amplify the 3’ flank. Primer extensions containing the restriction sites are lowercase bold type; introduced restriction sites are underlined.
Figure 1. Split-marker strategy for gene deletion. (A) Primers F1/F2 and F3/F4 amplify target gene flanking sequences. Primers M13R/HY and M13F/YG amplify "HY" and "YG" marker fragments, respectively. Note that primers F2 and F3 are hybrid, the 5’ ends are complementary to the M13F and M13R sequences, respectively, and the 3’ ends are gene specific. (B) Two separate PCR reactions (F1/YG) and (HY/F4) fuse the flank sequences to the 5’ (HY) or 3’ (YG) portions of HYG. (C) Homologous recombination and gene deletion. The two fusion PCR fragments are used directly for transformation. Homologous recombination between the overlapping regions of the selectable marker (HYG), and between the flank regions and chromosomal DNA results in a directed deletion.
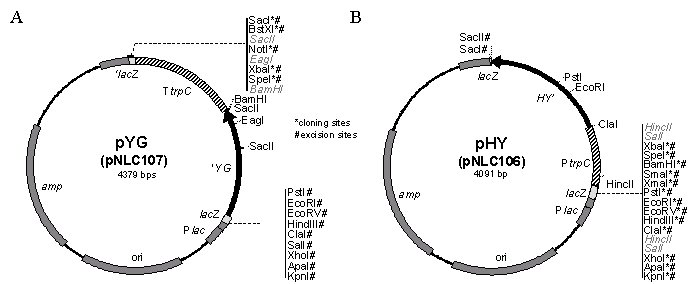
Figure 2. Split-marker plasmids. (A) pYG (pNLC107) is the 1426 bp XbaI-PstI fragment containing the 3’ end of the hygB gene from pUCATPH, Klenow treated to blunt the XbaI site, and cloned into pBLUESCRIPT (KS-) digested with SmaI and PstI. (B) pHY (pNLC106) is the 1143 bp XbaI-SacII fragment containing the TrpC promoter region and 5’ end of the hygB gene from pUCATPH cloned into XbaI-SacII digested pBLUESCRIPT. The overlap between the "HY" and the "YG" marker sequences is 445 bp. Restriction sites for cloning of flanks and excision of deletion constructs are noted with * and #, respectively.