Fungal Genet. Newsl. 50:15-16
A simple acetone-drying protocol was adopted to replace the lyophilization step while isolating genomic DNA from Aspergillus mycelia. This DNA is suitable for PCR, restriction enzyme digestion and Southern blot analysis with digoxigenin-labeled DNA probes. Acetone drying/ preservation can be a useful method in the molecular analysis of fungal DNA samples.
Many isolation procedures are available to obtain good quality genomic DNA, directly usable in molecular biology techniques (Zhang et al. 1996 FEMS Microbiol. Lett. 145:261-265; Min et al. 1995 Anal. Biochem. 225:94-100). These DNA isolation methods often employ grinding of either frozen (N. Talbot in Molecular and Cellular Biology of Filamentous Fungi, 2001, Oxford Univ. Press, p.23-31; Chow and Kafer 1993 Fungal Genet. Newsl. 40:25-27) or lyophilized (Al-Samarrai and Schmid 2000 Lett. Appl. Microbiol. 30:53-56) mycelia, before extraction with phenol. Lyophilized mycelia offer advantages in terms of storage and the quality of DNA isolated. However freeze-drying requires the equipment, relatively large amounts of starting material and up to a day to obtain dry mycelial powder. Acetone drying/ preservation is a successful practical method for molecular analysis of insect DNA samples (Fukatsu 1999 Molec. Ecol. 8:1935-1945). We have adopted and compared this technique as an effective alternative to freeze-drying of Aspergillus mycelia.
Experimental Procedures: Aspergillus niger (NCIM 565) and Aspergillus nidulans (NCIM 1211) were grown on complete medium (yeast extract, 0.6%; casein hydrolysate, 0.6% and dextrose, 1.0%; pH adjusted to 6.0 with 0.1M HCl). Cells were harvested by filtration, washed successively with cold sterile water and cold acetone (10 ml acetone per gram wet weight of mycelia; 1:10, w/v) on a Buchner funnel. The shrunken mycelial mat was held for 3h under cold acetone (1:10, w/v) at –20 C. The mat was transferred to fresh cold acetone (1:10, w/v) and stored for a further 3h at –20 C. The dehydrated mycelia so obtained were desiccated under vacuum (to remove acetone vapors) for 20 min. The dried sample was either directly used or stored at –20 C for future use. Mycelia grown on complete medium were also harvested, freeze-dried and stored at –20 C.
One gram of mycelium (either acetone-dried or lyophilized) was weighed out, ground with acid-washed sand (1.0 g) in a mortar with 3 ml of Lysis buffer (10 mM Tris-HCl pH 7.5, 0.5M NaCl and 1% SDS). To this, 4 ml of phenol:chloroform: isoamyl alcohol (PCI, 25:24:1) were added, mixed thoroughly and the suspension was taken up in Corex® tubes. After incubation at 65 C for 20 min, the suspension was centrifuged (at 4 C for 30 min at 4500 g in SW rotor). The aqueous supernatant was re-extracted once more with PCI (4:5, v/v). The aqueous phase was collected and gently mixed (by inverting) with 0.54 volume of isopropanol. The DNA precipitate was collected by centrifugation (at 4500 g) and the pellet washed once with cold 70% ethanol; It was carefully rinsed with cold, absolute ethanol and dried (30 min) in laminar hood. The dry DNA pellet was incubated under 1.5 ml of TE (Tris-HCl pH 8.0, 1.0 mM EDTA) containing RNase A (60 ug), at room temperature, for 15 min with occasional gentle wiggling. The DNA from the pellet is gradually extracted, making the TE translucent with DNA, while most of the polysaccharide is left behind. After centrifugation at 4500 g, aliquots of DNA solution (typically 300 ul) were ethanol precipitated and dissolved in 400 ul of TE or water, as required. We have isolated genomic DNA from acetone-dried mycelial preparations of A. niger and A. nidulans. The acetone drying protocol was contrasted with more familiar lyophilization of fungal mycelia, in preparing genomic DNA (Figure 1). While the DNA yields were comparable (0.4-0.5 micrograms per g of mycelial wet weight), acetone-dried samples gave DNA of good quality and predominantly of high molecular weight. This DNA served well as a template for PCR (Figure 1). To further confirm the quality of genomic DNA isolated, DNA obtained from the two methods were subjected to restriction enzyme (EcoRI) digestion and agarose gel electrophoresis followed by a transfer to Amersham Hybond N membrane. The Southern blot was hybridized with digoxigenin-labeled (DIG kit, Roche Diagnostics) A. niger arginase probe. Results shown in Figure 2 (a 5.0 kb A. niger DNA fragment hybridized to the DIG probe) indicate that genomic DNA isolated from acetone-dried mycelia is of good quality and suitable for use in molecular analysis. Our study was initially prompted by a temporarily nonfunctional freeze-dryer. The acetone-drying protocol (Fukatsu 1999 Molec. Ecol. 8:1935-1945) can effectively substitute for the lyophilization step; acetone-dried Aspergillus mycelia preserve well for future extraction of quality genomic DNA. Procedures to obtain mycelial acetone powders are reported (Munkres 1962 FGSC Protocols 1:12; Aguirre and Hansberg 1988 Fungal Genet. Newsl. 35:5-6). These acetone-drying-grinding protocols, although initially meant for protein/ enzyme work, could also be explored for isolating fungal genomic DNA. (We are grateful to DBT and BRNS-DAE for financial support)
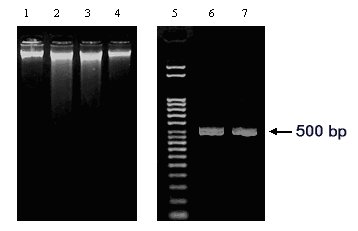
Figure 1: Genomic DNA isolated from two Aspergilli. The DNA samples prepared from acetone-dried (Lane 1: A. niger and Lane 4: A. nidulans) or freeze-dried (Lane 2: A. niger and Lane 3: A. nidulans) mycelia are shown. A. niger arginase gene (GenBank Acc. No. AF242315) specific primers were used to amplify A. niger genomic DNA. The PCR product (499 bp) corresponding to the template DNA in Lane 1 and Lane 2 are shown in Lanes 6 and 7, respectively. Lane 5: DNA ladder (Direct Load, Sigma)
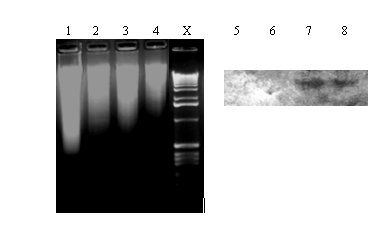
Figure 2: Digestion of genomic DNA with EcoRI and Southern blot analysis. Genomic DNA isolated from A. nidulans
(Lane 1: acetone-dried and Lane 2: freeze-dried) and A. niger (Lane 3: acetone-dried and Lane 4:
freeze-dried) mycelia are shown. Lanes 5,6,7 & 8 indicate the Southern blot (corresponding to Lanes 1,2,3 & 4,
respectively) analysis of the EcoRI digests hybridized with A. niger arginase (0.68 kb Xho-Sal fragment, AF242315)
specific, DIG-labeled probe. Lane X: DNA kb ladder (GIBCO BRL)